
合生元对慢性功能性便秘患者肠道特定菌群的影响及其功能注释
2019-01-16 08:38黄林生屈潇孔程潘登登张晓辉沈通一秦环龙
中国全科医学 2019年3期
黄林生 ,屈潇 ,孔程 ,潘登登 ,张晓辉 ,沈通一 ,秦环龙 *
随着生活条件和饮食结构的改变,慢性便秘的发病率呈逐渐上升趋势,慢性功能性便秘(CFC)在各年龄段人群中均有发生,特别是在老年人群中发病率更高[1]。目前便秘的临床治疗有传统药物治疗(膨松剂、泻剂、粪便软化剂、栓剂、促动力药、促肠液分泌剂)、灌肠、骨盆肌肉训练(生物反馈治疗)和手术治疗[2],然而微生态治疗目前已逐渐作为一种新的选择,如益生菌[3]、益生元[4]、合生元[5]和粪菌移植(FMT)[6]等方法。
本课题组前期的研究结果已经证实CFC与肠道菌群紊乱存在关联,采用合生元进行治疗不仅能够起到较好的临床效果,还能改变肠道菌群的结构,促进肠道微环境的改善[7]。目前乳酸杆菌和双歧杆菌被认为是传统的益生菌,其丰度水平下降被认为是疾病和老龄化的标志[8],而微生态制剂以这两种益生菌为主要成分,因此本研究将进一步检测相关益生菌菌群的丰度水平,同时基于前期的研究结果进一步行代谢相关的功能注释,从而探讨CFC的发病机制及微生态治疗的可能机制。
1 对象与方法
1.1 研究对象 2016年7月—2017年3月,选取本课题组前期纳入的24例CFC患者[7],根据送检粪便样本量及16S-rRNA结果剔除离群值,最终筛选出6例CFC患者。6例患者均采用合生元(商品名谊畅)进行治疗,每条制剂(长双歧杆菌、嗜酸乳杆菌、植物乳杆菌、乳双歧杆菌、婴儿双歧杆菌以及低聚木糖)包含1×1010CFU/g的益生菌。服用方法为饭后30 min温水送服,3次/d,持续治疗1个月。同时在社区招募6例健康者作为健康组。健康组无CFC及其他慢性疾病,纳入前未服用益生菌和抗生素等药物。本研究经同济大学附属第十人民医院伦理委员会审核(SHSY-IEC-3.0/16-53/01)通过。患者均知情同意。研究对象的基本信息见表1。
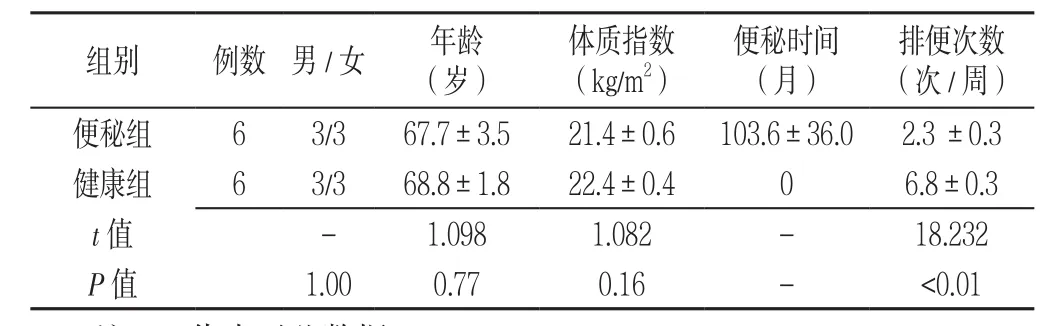
表1 研究对象的基本信息Table 1 Baseline data of the participants
1.2 菌群检测及分析 便秘组患者于门诊留取新鲜粪便样本,给予合生元治疗1个月后再次收集粪便样本。同时在社区留取健康组的粪便样本,与冰块一起运送至实验室并存放于-80 ℃冰箱储存待检测。(1)DNA抽提:依据粪便样本类型使用相应的试剂盒抽提DNA。(2)DNA质检:使用1%琼脂糖凝胶电泳检测抽提的基因组DNA并检测DNA水平。(3)DNA片段化:将DNA打断成约500 bp的片段。(4)文库构建:DNA片段末端修复和连接A碱基,在DNA片段两端连接接头,琼脂糖凝胶电泳进行片段筛选,PCR扩增和纯化,Agilent 2100检测文库大小,qPCR检测文库摩尔浓度,NaOH变性,产生单链片段。(5)Cluster簇生成:DNA片段的一段与引物碱基互补,固定在芯片flow cell上,另一端随机与附近另一个引物互补,并进行固定,随后进行PCR扩增,产生DNA簇,再线性化成为单链。(6)Illumina HiSeq测序,在flow cell加入DNA聚合酶和4种荧光标记的Dntp,每次循环只掺入单种碱基,激光扫描芯片,捕捉荧光信号,读取核苷酸种类。重复上述过程,依次统计每轮收集到的荧光信号结果,获知模板DNA片段序列。检测并计算各菌群的丰度水平。
1.3 生物信息分析 (1)基于KEGG数据库的功能注释统计:运用KEGG图形功能来表达各个代谢途径及各途径之间的关系,同时将不同的代谢途径进行分类并对存在联系的相关代谢途径进行关联。最后运用BLAST将检测出的基因集与KEGG的基因数据库进行比对,获得KEGG数据库的KO注释信息。(2)KO是蛋白质(酶)的一个分类体系,将序列高度相似并且在同一条通路上有相似功能的蛋白质归为一组。根据KEGG数据库得到的KO注释信息,累加对应的基因丰度,得到KO丰度水平。(3)KO的主成分分析(PCA):将数据变换到一个新的坐标系统中,通过对数据的降维保持数据集对方差贡献最大的特征。第一大方差在第一个坐标(称为第一主成分)上,第二大方差在第二个坐标(第二主成分)上。
1.4 统计学方法 使用SPSS 17.0统计软件进行数据分析。符合正态分布的计量资料采用(±s)表示,两组间比较采用成组t检验。计数资料的比较采用Fisher's确切概率法。菌群丰度水平治疗前后的比较采用配对样本的Wilcoxon秩和检验,两独立样本比较均采用非参数检验(Mann-Whitney U 秩和检验)。以P<0.05为差异有统计学意义。
2 结果
2.1 传统益生菌的丰度水平比较 10种双歧杆菌在种水平被检出,其中健康组B.dentium丰度水平高于便秘组治疗前,便秘组治疗前B.adolescentis、B.animalis丰度水平高于健康组,差异有统计学意义(P<0.05,见表2)。便秘组治疗后B.animalis丰度水平较便秘组治疗前增高,差异有统计学意义(P<0.05,见表3)。
38种乳酸杆菌被检出,其中健康组L.oris、L.reuteri丰度水平高于便秘组治疗前,差异有统计学意义(P<0.05,见表2)。便秘组治疗后L.oris丰度水平较治疗前增高,差异有统计学意义(P<0.05,见表3)。
合生元中补充的益生菌(长双歧杆菌、嗜酸乳杆菌、植物乳杆菌、乳双歧杆菌、婴儿双歧杆菌)丰度水平在治疗前后变化不明显,本研究未列出具体数据。
埃希菌属共计8种菌群被检出,其中Escherichia_coli(0.022与 0.001,Z=-2.201,P=0.028) 经 过 合 生元治疗后丰度降低。普氏菌属(Prevotella)共计33种菌种被检出,其中便秘组治疗前P-denticola(0与1.85×10-6,Z=-2.286,P=0.028),P-melaninogenica(4.43×10-8与 1.09×10-6,Z=-2.137,P=0.040),P-multiformis(0 与 1.39×10-5,Z=-2.678,P=0.028),P_s_MSX73(1.6310-7与 1.34×10-5,Z=-2.419,P=0.015),P_s_oral_taxon_317(0与 2.12×10-7,Z=-2.286,P=0.028),P_s_oral_taxon_472(0 与3.16×10-7,Z=-2.286,P=0.028)6种菌群丰度水平健康组,而便秘组治疗前Prevotella_s_C561菌(1.51×10-5与3.41×10-6,Z=-2.242,P=0.026)丰度水平高于便秘组治疗后,差异有统计学意义。
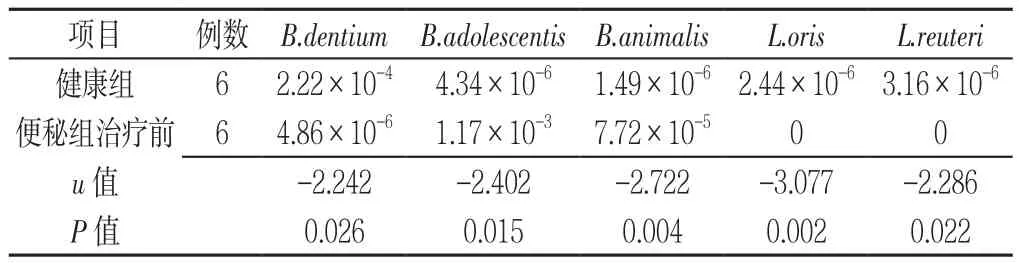
表2 健康组和便秘组治疗前双歧杆菌和乳杆菌丰度水平比较Table 2 Comparison of the abundance of Bifidobacterium and Lactobacilli between the healthy group and constipation group before treatment

表3 便秘组治疗前、后双歧杆菌的丰度水平比较Table 3 Comparison of the Bifidobacterium and Lactobacillus abundance in constipation group before and after treatment
2.2 基因功能注释 对健康组、便秘组治疗前、便秘组治疗后的所有菌群数据进行KEGG功能预测注释,肠道菌群基因和碳水化合物、氨基酸的代谢十分相关,基因所占比例超过10%(见图1)。根据KEGG数据库得到的KO注释信息,累加对应的基因丰度,选取所用样品中丰度排名前30的KO及其在每个样品中的丰度信息绘制热图。通过对3组样本的KO集进行热图分析对比发现KO2014在便秘组治疗前、后含量均较高,即葡萄糖dTDP-4、6-脱水酶在便秘组治疗前后的丰度水平较高(见图2)。而通过对3份样本的KO集进行PCA分析,发现便秘组治疗前、后与健康组菌群KO集差异明显,而便秘组治疗前、后差异不明显。其PCA1值为27.47%,PCA2值为12.01%(见图3)。
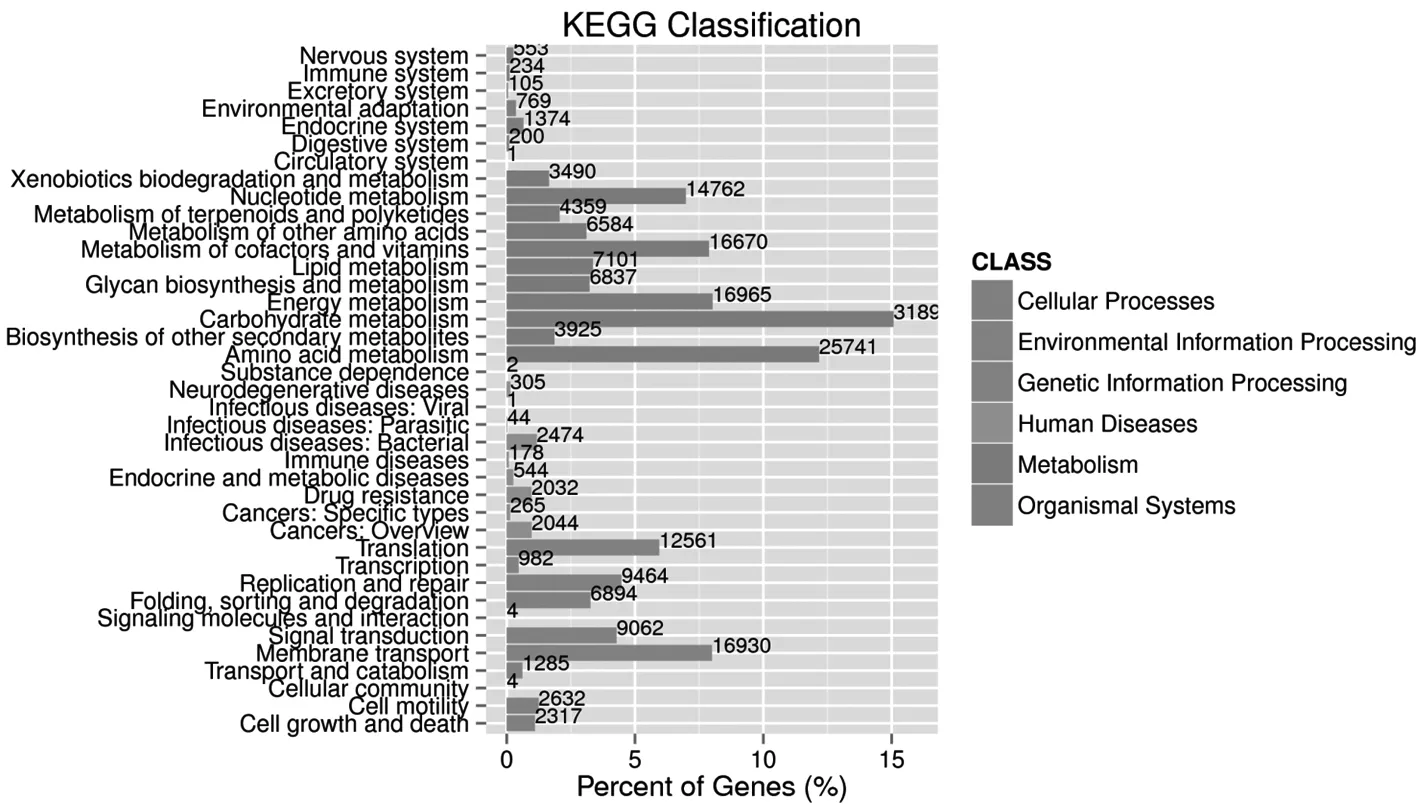
图1 KEGG注释结果分类统计Figure 1 Classification statistical results of KEGG annotation
3 讨论
最近慢性便秘的微生态治疗受到越来越多的关注,但目前国内外研究关注较多的是微生态治疗的临床效果,缺乏对微生态治疗对肠道微环境影响的进一步分析[9-10]。双歧杆菌和乳酸杆菌一直被认为是传统的益生菌,部分已知的种类一直被广泛应用于益生菌产品中,L.reuteri DSM 17938在预防婴幼儿便秘、增加排便次数和降低便秘带来的医疗费用增加方面具有积极的作用[11],另一综述证明L.reuteri在儿童中应用是安全和有效的[12]。但益生菌应用于CFC的科学性还有待进一步研究。
在前期研究基础上,本研究通过宏基因组测序和分析,对便秘组治疗、前后的肠道菌群行进一步鉴定。通过比较发现双歧杆菌中健康组B.dentium丰度水平高于便秘组治疗前,而便秘组治疗前B.adolescentis和B.animalis丰度水平高于健康组,便秘组治疗后B.animalis丰度水平较治疗前增加。乳酸杆菌中健康组L.oris和L.reuteri丰度水平高于便秘组治疗前。Prevotella菌中6种菌属健康组丰度水平高于便秘组治疗前。国外相关试验也有不同的发现,KIM等[13]发现Bifidobacterium和Bacteroides菌群丰度水平在CFC患者的粪便中是降低的,而B.longum在便秘儿童中的丰度水平较高[14]。ZHU等[15]发现Prevotella菌属在便秘人群中的丰度水平是降低的,本研究结果与之相似。

图2 健康组、便秘组治疗前、便秘组治疗后KO集热图比较Figure 2 Heatmap of KO in the samples of healthy group,constipation group before treatment,constipation group after treatment
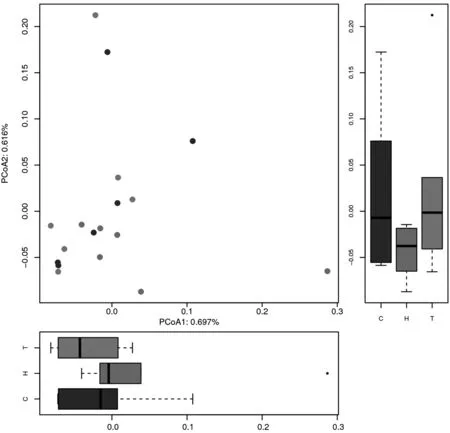
图3 健康组、便秘组治疗前、便秘组治疗后KO的主成分分析结果Figure 3 PCoA of KO in the samples of healthy group,constipation group before treatment,constipation group after treatment
在研究慢性便秘人群的肠道菌群结构及采用微生态制剂进行治疗时需考虑到年龄因素对结果的干扰。B.longum、B.breve和B.bifidum在婴幼儿中处于优势菌群,而B.catenulatum、B.adolescentis和B.longum在成年人群中处于优势地位[8]。另一项综述研究发现B.lactis,而非L.casei作为益生菌治疗时能够显著降低肠道传输时间,恢复粪便的性状和增加排便次数[3]。一项meta分析发现益生菌治疗能够增加排便次数,分组研究显示在亚洲人群的治疗中只能增加排便次数,但对粪便的性状没有明显改善[16]。因此并不是所有的传统益生菌均适用于对慢性便秘的治疗,通过对特定年龄的肠道菌群分析,并基于严格的菌群结构特定进行针对性的治疗甚至是进行个性化的治疗才是未来微生态研究的重点方向。
本研究通过对肠道菌群功能预测发现肠道菌群与肠道中的碳水化合物、氨基酸等物质的代谢相关,但未能发现在CFC治疗中发挥重要作用的相关基因,而代谢组学的研究发现益生菌对肠道功能的改善不仅体现在对菌群的优化[17],还在于对乳酸、短链脂肪酸[18]和5羟色胺(5-HT)[19-20]等肠道代谢物的改变。
目前相关的动物实验结果提示菌群产生的长链饱和脂肪酸(十七烷酸和硬脂酸)可促进大鼠结肠肌肉收缩、增加排便频率,而且与普氏菌属、乳杆菌属和别样杆菌的丰度水平增高相关[21],而肠道中色氨酸通过肠嗜铬细胞色氨酸羟化酶1(TpH1)代谢产生5-HT,从而发挥刺激肠道运动、促进肠-脑轴信号等功能[22],单胺与5-HT类似,通过对小鼠定殖可产生色胺的工程多形拟杆菌,可促进胃肠道蠕动,这些研究为CFC的治疗提供了更好的思路[23]。目前使用合生元治疗CFC患者具有较好的临床效果[24],而基于肠道菌群研究的基础上[25],通过对菌群和代谢物质的关联分析,才能在未来的治疗中提供精准化的治疗方案。
本研究在细菌种水平的角度揭示传统益生菌在CFC以及在合生元治疗中的改变情况,对菌群的定位更加精确,发现了不同益生菌在合生元治疗中可能存在的意义,为实现CFC的精准化治疗提供依据。但本研究的不足之处在于样本量还需进一步扩大,同时还需从粪便代谢组学方向对临床样本进行后续的验证。
作者贡献:黄林生进行文章的构思与设计,撰写论文并对论文进行修订;沈通一进行研究的实施与可行性分析;孔程进行数据收集;潘登登进行数据整理;张晓辉进行统计学处理;屈潇进行结果的分析与解释;沈通一、秦环龙负责文章的质量控制及审校;秦环龙对文章整体负责,监督管理。
本文无利益冲突。
猜你喜欢
中老年保健(2022年2期)2022-08-24
世界科学技术-中医药现代化(2022年2期)2022-05-25
当代水产(2022年3期)2022-04-26
中国饲料(2022年5期)2022-04-26
昆明医科大学学报(2022年3期)2022-04-19
中国乳品工业(2022年3期)2022-04-11
当代水产(2021年2期)2021-03-29
保鲜与加工(2021年1期)2021-02-06
乳业科学与技术(2020年4期)2020-12-10
消费者报道(2019年3期)2019-06-12
