
PF-4708671和NVP-BEZ235联合用药协同抑制MDA-MB-436和A549细胞增殖
2019-01-08 06:07:12彭培佩晨3李山虎
中国药理学与毒理学杂志 2018年8期
彭培佩*,王 健*,陈 晨3,,黄 芳,王 芃,李山虎,王 辉
(1.广东药科大学生命科学与生物制药学院,广东省生物技术候选药物研究重点实验室,广东广州 510006;2.军事科学院军事医学研究院生物工程研究所细胞工程实验室,北京 100850;3.河南科技大学医学院,河南洛阳 471023;4.北京城市学院生物医药学部,北京 100083)
p70核糖体S6激酶β-1(ribosomal protein S6 kinase beta-1,S6K1)属于蛋白激酶A/蛋白激酶G/蛋白激酶C家族的丝氨酸/苏氨酸激酶,参与调节西罗莫司(雷帕霉素)复合物1〔mammalian target of sirolimus(Rapamycin)complex 1,mTORC1〕蛋白激酶下游的蛋白质合成和细胞生长[1-2],而作为40S核糖体亚基组成部分的S6蛋白是S6K1最重要的底物之一[3]。通常认为,mTORC1-S6K1信号轴调控细胞转录、翻译、蛋白质合成、脂质合成、细胞生长和细胞代谢等过程。因此,S6K1在包括肥胖、糖尿病、衰老和癌症等许多疾病中起重要作用,研究者已提出通过抑制该激酶活性来治疗癌症和胰岛素抗性的策略[4-5]。
PF-4708671是S6K1的特异性抑制剂,通过诱导依赖于mTORC1的S6K1的T环和疏水基序的磷酸化,进而抑制S6K1底物S6蛋白的磷酸化[6]。研究表明,PF-4708671能抑制线粒体复合物Ⅰ,导致线粒体功能异常,并诱导过量的活性氧簇(reactive oxygen species,ROS)产生,通过激活胱天蛋白酶或释放细胞色素c,最终导致细胞死亡[6-8]。另外,最近的研究还发现,PF-4708671也可通过减少抗凋亡蛋白的表达,诱导对他莫昔芬耐药的MCF-7细胞死亡[9-11]。
磷脂酰肌醇-3-激酶(phosphatidylinositol-3-kinase,PI3K)/蛋白激酶B(protein kinase B,AKT)和mTOR信号通路是细胞内重要的传导通路之一,与细胞生长、增殖和凋亡密切相关[12]。PI3K/AKT/mTOR信号通路在多种癌症中被激活,其激活机制包括肿瘤抑制因子磷酸酶和张力蛋白同源性(phosphatase and tensin homolog,PTEN)基因功能的丧失、PI3K的扩增或突变、AKT的扩增或突变和生长因子受体的激活等[13]。一系列临床研究均表明,mTOR激酶抑制剂(如西罗莫司及其衍生物依维莫司等)可用于治疗如食管鳞状细胞癌、肺癌、肾细胞癌和前列腺癌等实体瘤,而许多PI3K/AKT/mTOR信号分子的靶向性药物也正处于临床前开发或早期临床试验阶段[13-14]。但临床试验也发现,mTOR激酶抑制剂西罗莫司和依维莫司在抑制S6K1活性的同时能反馈激活AKT信号通路,继而通过调控Bcl-2、叉头框蛋白家族因子、凋亡蛋白抑制因子(inhibitor of apoptosis proteins,IAP)、胱天蛋白酶9、糖原合酶激酶3(glycogen synthase kinase 3,GSK-3)、结节性硬化症复合物2(tuberous sclerosis complex 2,TSC-2)等分子活性抑制肿瘤细胞凋亡、促进增殖等作用减弱治疗效果[15-17]。为此,本研究通过探索S6K1抑制剂PF-4708671调控PI3K/AKT/mTOR信号通路的作用机制,提出与NVP-BEZ235联合用药的策略,以期对肿瘤靶向性治疗临床用药提供有益的线索。
1 材料与方法
1.1 细胞、药物、试剂和主要仪器
感受态细胞大肠杆菌DH5α、人乳腺癌细胞MDA-MB-436和肺癌细胞A549购自美国ATCC;西罗莫司(sirolimus)、PF-4708671和NVP-BEZ235购于美国Selleck公司;山羊抗微管蛋白(γ-Tubulin)多克隆抗体购自美国Santa Cruz公司;兔抗磷酸化AKT473(p-AKT473),p-S6,AKT和S6K1多克隆抗体均购于美国Cell Signaling公司;山羊抗兔和兔抗山羊IgG抗体购于北京中杉金桥生物公司;DMEM高糖培养基和胰蛋白酶购于美国Gibco公司;二甲亚砜(DMSO)购于美国Sigma公司;胎牛血清购于杭州四季青公司;青霉素和链霉素购于华北制药有限公司;嘌呤霉素购于美国HyClone公司;蛋白酶抑制剂和甲紫(结晶紫)购于美国Amresco公司;BCA蛋白定量试剂盒、CO2恒温培养箱和MULTISKAN FC酶标仪均购自美国Thermo公司;ECL发光液购于北京天根生化科技有限公司;Cell Counting Kit 8(CCK8)试剂盒购于东仁化学科技有限公司;Lipo3000试剂盒购于赛默飞世尔中国;5417R低温高速离心机及5418台式室温离心机购于德国Eppendorf公司;电泳槽和湿转膜仪购于美国Biorad公司。
1.2 细胞培养
MDA-MB-436和A549细胞均用含10%胎牛血清、青霉素100 kU·L-1和链霉素0.1 g·L-1的DMEM高糖培养基在37℃,5%CO2的培养箱中培养。待细胞生长密度长到80%~90%时进行传代培养。
1.3 质粒构建和转化
根据相关文献[18],利用PLKO.1载体设计干扰S6K1表达的短发夹RNA(sh-S6K1),引物序列为F:CC⁃GGGCATCGGCACCACTTCCAATACTCGAGTATT⁃GGAAGTGGTGCCGATGCTTTTTG;R:AATTCAAAAAGCATCGGCACCACTTCCAATACTCGAGTATTGGAAGTGGTGCCGATGC。引物由北京赛百盛生物工程公司合成,4℃保存。使用PLKO.1载体按胶回收试剂盒(Axygen公司)说明书进行酶切回收,将设计的短序列进行退火连接,将载体片段和退火片段通过T4连接酶进行连接,将连接产物利用大肠杆菌DH5α进行转化,涂氨苄平板,挑单克隆摇菌,按质粒提取试剂盒(Axygen公司)说明书提取质粒,并测定浓度,-20℃保存。送生物工程(上海)股份有限公司北京测序部得到测序结果。按照Lipo3000试剂盒说明书操作,获得稳定敲低S6K1表达的MDA-MB-436和A549细胞。
1.4 Western蛋白印迹法检测p-AKT473,AKT和S6K1蛋白水平
在接种MDA-MB-436和A549细胞的培养皿内加入终浓度为30 μmol·L-1的PF-4708671,处理0,1,3,6,12和24 h后收取细胞;以及单独或联合加入PF-4708671(30 μmol·L-1)或西罗莫司(10 nmol·L-1)处理24 h后收取细胞;向接种敲低S6K1的上述2种细胞的培养皿中加入终浓度为30 μmol·L-1的PF-4708671,处理24 h后收取细胞;以及单独或联合加入 PF-4708671 30 μmol·L-1或 NVP-BEZ235 100 nmol·L-1,处理24 h后收取细胞;用含蛋白酶抑制剂和磷酸酶抑制剂的RIPA裂解液裂解细胞,利用BCA比色法在酶标仪570 nm处检测吸光度(A570nm)值,测定标准曲线得出蛋白样品的浓度。向蛋白样品中加入5×SDS加样缓冲液并煮沸8 min,对蛋白进行预变性处理。以10%SDS-PAGE聚丙烯酰胺凝胶电泳分离蛋白样品后,在湿转装置中以200 mA进行恒流转膜2.5 h,在5%脱脂奶粉中封闭约30 min。使用5%的脱脂奶粉稀释一抗(p-AKT473,AKT和S6稀释比例为1∶1000,p-S6稀释比例为1∶3000),在4℃的摇床上孵育过夜。用TBST洗膜3次后加入二抗(稀释比例1∶5000)室温孵育6 h,之后用TBST洗膜3次,每次5 min,以ECL法显影及定影。胶片经扫描后用Image J进行积分吸光度分析。
1.5 CCK-8法检测细胞存活
将处于对数生长期的上述2种细胞消化后,按每孔分别3500和2500细胞接种到96孔板中,每孔重复3次,置于CO2培养箱24 h后,各孔单独加入0,10,20,30和40 μmol·L-1的PF-4708671或联合加入 NVP-BE235(MDA-MB-436:8 nmol·L-1;A549:1 μmol·L-1)抑制剂72 h后去除原孔内培养液,每孔加入100 μL稀释后的CCK8工作液(按CCK8原液与培养液1∶9的比例配制)继续孵育2 h,而后利用酶标仪在450 nm处检测吸光度(A450nm)值,细胞存活率(%)=(加药组A450nm-空白组A450nm)/(正常对照组A450nm-空白组A450nm)×100%。实验重复3次。
1.6 平板克隆形成实验检测细胞增殖
将处于对数生长期的上述2种细胞按每孔3000细胞接种到6孔板内,待孔内细胞克隆生长到一定大小时,单独或联合加入PF-4708671(30 μmol·L-1)和NVP-BEZ235(100 nmol·L-1),6 d后去除原培养液,加入PBS洗涤3次,加入甲醇固定细胞层约30 min,加入甲紫染液染色30 min,用流水缓慢清洗孔内剩余染料后放置于室内风干。显微镜下计数≥10个细胞的克隆数。实验重复3次取平均值。
1.7 统计学分析
计量资料以±s表示。使用SPSS19.0进行数据处理和统计学分析,Studentt检验(或校正t检验)用于两组间比较;单因素方差分析进行多组比较,P<0.05为具有统计学意义。应用Chou-Talalay方法分析联合用药后的联合指数(combination index,CI),定量描述联合用药的相加作用(CI=1)、协同作用(CI<1)以及拮抗作用(CI>1)。
2 结果
2.1 PF-4708671对MDA-MB-436和A549细胞中p-S6及AKT表达水平的影响
Western蛋白印迹结果(图1)显示,PF-4708671 30 μmol·L-1作用于上述2种细胞后,p-S6蛋白表达水平显著降低(P<0.01),而p-AKT473蛋白表达水平显著上升(P<0.01),同时AKT总蛋白表达水平无明显差异(图1A,C)。表明S6K1抑制剂PF-4708671抑制上述2种细胞中S6磷酸化水平的同时能反馈激活AKT活性。
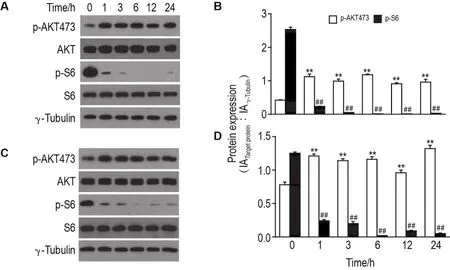
Fig.1 Effect of PF-4708671(PF)on protein expression levels of protein kinase B(AKT),p-AKT473,S6 and p-S6 in MDA-MB-436(A,B)and A549(C,D)cells by Western blotting.Cells were treated with PF 30 μmol·L-1for 0,1,3,6,12 and 24 h,respectively.B and D was the semi-quantitative analysis of A and C,respectively.IA:integrated absorbance.±s,n=3.**P<0.01,compared with p-AKT473 in 0 h group;##P<0.01,compared with p-S6 in 0 h group.
2.2 PF-4708671和西罗莫司联用对MDA-MB-436和A549细胞中p-S6及AKT表达水平的影响
Western蛋白印迹结果(图2)显示,单用或联用mTOR抑制剂西罗莫司10 nmol·L-1和PF-4708671 30 μmol·L-1作用上述2种细胞,均能抑制p-S6活性并导致AKT反馈激活(P<0.01);当两者联用时,对p-S6活性有进一步的抑制(P<0.01),且进一步激活了AKT活性(P<0.01),AKT总蛋白表达水平无明显差异(图2A,C)。表明在上述2种细胞中,西罗莫司和PF-4708671在抑制S6K1活性的同时都能导致AKT反馈激活,且具有叠加作用。
2.3 PF-4708671对敲低S6K1的MDA-MB-436和A549细胞中S6K1及AKT表达水平的影响
Western蛋白印迹结果(图3)显示,敲低S6K1后,ATK活性相较于野生型细胞明显上升(P<0.01);用PF-4708671作用野生型细胞时,能导致p-AKT473表达水平显著增加(P<0.01),但用PF-4708671作用于敲低S6K1后的上述2种细胞时,其对p-AKT473的表达水平增加的程度明显低于PF-4708671作用野生型细胞中p-AKT473增加的水平(P<0.01)(图3E),同时AKT总蛋白表达水平无明显差异(图3A,C)。这一研究表明,敲低S6K1后的确能显著增加p-AKT473表达水平,同时PF-4708671可通过抑制S6K1激活AKT活性。
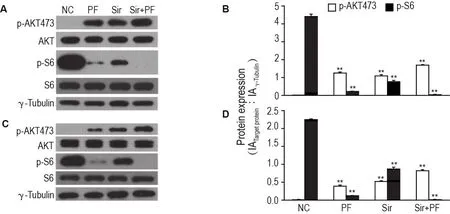
Fig.2 Effect of PF combined with sirolimus(Sir)on protein expression levels of p-AKT473,AKT,S6 and p-S6 in MDA-MB-436(A,B)and A549(C,D)cells by Western blotting.Cells were treated with PF(30 μmol·L-1,24 h)and Sir(10 nmol·L-1,24 h)alone or in combination.B and D was the semi-quantitative analysis of A and C,respectively.±s,n=3.**P<0.01,compared with corresponding normal control(NC)group.
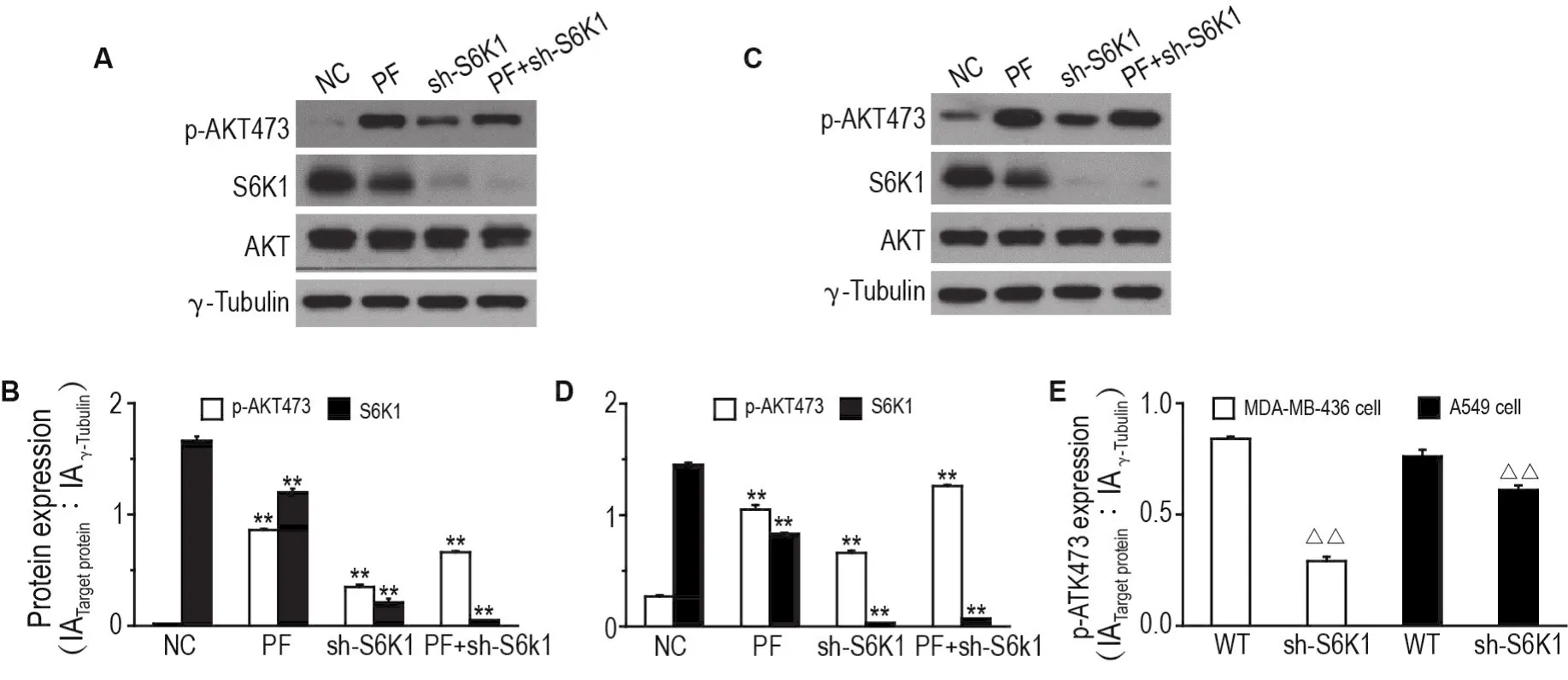
Fig.3 Effect of PF on ribosomal protein S6 kinase beta-1(S6K1)and p-AKT473 expression levels in MDA-MB-436(A,B)and A549(C,D)cells after S6K1 was knocked down by small interfering RNA(sh-S6K1)by Western blotting.The sh-S6K1 cells were treated with PF 30 μmol· L-1for 24 h.B and D was the semi-quantitative analysis of A and C,respectively.E was the changes in p-AKT473 expression in wild-type(WT)and sh-S6K1 cells after being treated with PF,and was the semi-quantitative analysis of A and C.±s,n=3.**P<0.01,compared with corresponding NC group;△△P<0.01,compared with corre⁃sponding WT group.
2.4 PF-4708671联合NVP-BEZ235对MDA-MB-436和A549克隆形成和细胞存活的影响
Western蛋白印迹结果(图4)显示,PI3K/mTOR双重抑制剂NVP-BEZ235可显著抑制由PF-4708671所导致的p-AKT473活性增强(P<0.01)。平板克隆结果(图5A-D)显示,与单用PF-4708671或NVP-BEZ235相比,两者联合处理后MDA-MB-436及A549细胞的克隆形成数显著降低(P<0.01)。CCK8结果(图5E,F)表明,与正常对照组相比,单用PF-4708671及NVP-BEZ235均具有抑制肿瘤细胞生长的效果(P<0.01),但2种抑制剂联用时,显著增强了抑制肿瘤细胞生长的作用(P<0.01),且两药联用存在协同作用(CI<1)。

Fig.4 Effect of PF combined with NVP-BEZ235(BEZ)on expression levels of AKT,p-AKT473,S6 and p-S6 kinase in MDA-MB-436(A,B)and A549(C,D)cells by Western blotting.The cells were treated with PF(30 μmol·L-1,24 h)and BEZ(100 nmol·L-1,4 h).B and D was the semi-quantitative analysis of A or C,respectively.±s,n=3.**P<0.01,compared with corresponding NC group.
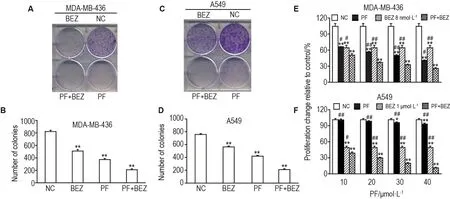
Fig.5 Effect of PF combined with BEZ on clonal formation and cell survival of MDA-MB-436(A,B,E)and A549(C,D,F).A and C:the cells were treated with PF(MDA-MB-436:20 μmol·L-1;A549:60 μmol·L-1)and BEZ(MDA-MB-436:8 nmol·L-1;A549:20 nmol·L-1)alone or in combination for cloning experiment respectively.B and D:count analysis of each hole for A and C.E and F:the cells were treated with the indicated concentrations gradient of PF with BEZ(MDA-MB-436:8 nmol·L-1;A549:1 μmol·L-1)alone or in combination for cell survival assay.±s,n=3.*P<0.05,**P<0.01,compared with NC group;#P<0.05,##P<0.01,compared with PF+BEZ group.
3 讨论
本研究发现,S6K1抑制剂PF-4708671和PI3K/mTOR激酶抑制剂NVP-BEZ235联用于乳腺癌细胞MDA-MB-436和肺癌细胞A549,可显著增强单用时对肿瘤细胞的生长抑制作用。
PI3K/AKT/mTOR信号通路与多种癌症的发生有关,因而被认为是靶向性治疗的有效靶点,但是常常由于受体酪氨酸激酶(receptor tyrosine kinases,RTK)信号增强,或和其他促生长信号通路之间的交互作用,导致耐药性和不良预后的发生[19-21]。
PI3K/AKT/mTOR信号通路激活下游的S6K1以调节细胞代谢、增殖[22]。有研究发现,激活的S6K1可抑制PI3K上游调控分子胰岛素受体底物1(insulin receptor substrate 1,IRS-1)的功能活性,导致PI3K/AKT通路无法被正常激活而表现出一种负反馈调控机制[2]。因此,当用西罗莫司抑制mTORC1会解除S6K1反馈抑制环路而导致AKT的持续激活,使得肿瘤细胞对西罗莫司产生耐药性,减弱其潜在的抗癌活性[23]。但也有研究对此结论提出了质疑。Wang等[24]研究发现,用S6K1抑制剂或者直接敲低S6K1表达都不能激活AKT活性,进而认为西罗莫司或mTORC1抑制剂不是通过解除IRS-1/PI3K/AKT信号通路的负反馈调控来激活AKT活性。而本研究的结果发现,S6K1抑制剂PF-4708671和西罗莫司具有相似的作用分子机制,都可以通过抑制S6K1活性来激活AKT活性。本研究首先发现,用S6K1抑制剂PF-4708671处理MDA-MB-436和A549细胞后抑制S6K1活性同时相应激活了AKT活性;而通过敲低肿瘤细胞中S6K1表达,可直接上调AKT磷酸化水平,说明S6K1直接参与了负调控ATK活性。同时发现,在敲低S6K1表达的突变型细胞中,PF-4708671依然能激活AKT活性;但相对于野生型细胞而言,PF-4708671对ATK的激活程度显著减弱,这都提示PF-4708671激活AKT活性的分子机制可能是通过抑制S6K1活性来实现的。
由于AKT的激活经常与细胞生存和抗癌治疗有关,因而认为PF-4708671作用下的AKT激活也会减弱PF-4708671的抗肿瘤作用。因此,本研究首先证实PF-4708671对AKT的反馈激活可被PI3K和mTOR双重ATP竞争性抑制剂NVP-BEZ235所阻断,在此基础上提出了联合用药的策略。研究结果证实,在PF-4708671和NVP-BEZ235联用时显著增强PF-4708671单用对肿瘤细胞的抑制作用。因此,对于PI3K/AKT/mTOR信号通路异常激活所导致的肿瘤,可通过联用S6K1抑制剂和PI3K/AKT抑制剂来增强靶向性治疗的效果。
猜你喜欢
天津医科大学学报(2021年3期)2021-07-21 09:03:46
世界科学技术-中医药现代化(2021年12期)2021-04-19 12:31:40
重庆医学(2019年8期)2019-04-25 13:15:54
中国抗生素杂志(2019年2期)2019-03-13 07:07:42
生物医学工程研究(2017年1期)2017-10-18 01:39:14
环球人物(2017年9期)2017-05-31 13:23:23
中国医药生物技术(2015年4期)2015-12-26 08:26:36
中国医学科学院学报(2013年6期)2013-03-11 20:26:04
