
氧化应激、炎症和自噬在脑缺血损伤中作用机制及治疗策略
2019-01-08 06:07张思然李成檀张丽慧
中国药理学与毒理学杂志 2018年8期
张思然,李成檀,杨 怡,张丽慧
(1.浙江医院神经内科,浙江杭州 310013;2.杭州师范大学医学院药学系,医学神经生物学杭州市重点实验室,浙江杭州 310036)
急性脑缺血是世界范围内重大的脑血管疾病,具有发病率和致残致死率高的特点,目前尚缺乏治疗或延缓脑缺血损伤的有效方法。缺血引起的脑组织缺氧以及缺血再灌注,触发了一系列脑缺血级联反应,最终导致神经元死亡和神经功能障碍[1-3]。近期大量研究表明,氧化应激、炎症和自噬作为脑缺血再灌注后的关键分子事件,在缺血性脑损伤的发生发展过程中起重要作用,靶向这些机制显示有益的神经保护作用。进一步研究发现,脑缺血后的氧化应激对细胞自噬和炎症反应有重要调控作用,反之,自噬活性改变又对氧化应激与炎症反应产生影响,三者之间存在密切而复杂的相互作用。而且,脑内神经元、胶质细胞和血管内皮细胞等构成神经血管单元(neurovascular unit,NVU)共同参与脑缺血损伤[2]。本文主要介绍脑缺血损伤过程中氧化应激、炎症和自噬的作用机制及其相互作用研究进展。在此基础上,进一步讨论脑缺血损伤的潜在治疗干预措施,旨在为寻找有效的神经保护策略提供新思路。
1 脑缺血后氧化应激损伤与抗损伤
机体氧化和抗氧化系统之间的稳态被破坏而形成的氧化应激状态是脑缺血损伤的关键机制[4-5]。体内活性氧(reactive oxygen species,ROS)主要通过细胞线粒体琥珀酸脱氢酶(succinate dehydro⁃genase,SDH)和NADPH氧化酶等途径产生。已有证据表明,脑缺血再灌注后选择性聚集的琥珀酸被SDH快速氧化,通过线粒体复合物Ⅰ逆向电子传递产生过量的线粒体ROS是脑缺血后ROS的重要来源[6-7]。ROS作为脑内关键的信号分子直接或间接介导缺血性脑损伤的诸多病理过程。主要机制有[2-8]:①直接破坏细胞脂类、蛋白质和核酸;②损伤细胞线粒体,促进细胞色素c等多种蛋白释放,进而诱发线粒体依赖的细胞凋亡;③损伤血管内皮细胞,增加血脑屏障(blood-brain barrier,BBB)通透性;④诱导转录因子NF-κB激活和炎症细胞因子产生;⑤调节自噬。另一方面,体内产生NO的一氧化氮合酶(nitric oxide synthase,NOS)存在3种亚型:内皮型NOS(endothelial NOS,eNOS)、神经元型NOS(neuronal NOS,nNOS)和诱导型NOS(inducible NOS,iNOS)。eNOS和nNOS为钙依赖型,调节产生低浓度的NO。eNOS通过NO的血管活性作用促进循环血流,产生神经保护作用[5,9]。脑缺血后nNOS在早期表达增加,通过与连接蛋白突触后致密物95(postsynaptic density-95,PSD-95)结合,介导谷氨酸兴奋性毒性[5,10]。小分子 ZL006抑制nNOS-PSD-95相互作用减轻缺血后神经元损伤,促进其修复[11-12]。iNOS为非钙依赖型,其活性主要由信号转导通路所调控,脑缺血诱导iNOS在炎症及神经细胞持续高表达并产生大量NO,在缺血性脑损伤中起重要作用。过量NO可进一步与超氧阴离子反应生成强氧化性的过氧亚硝酸盐,也可干扰超氧化物歧化酶(superoxide dismutase,SOD)活性,参与脑缺血损伤[4,9]。
在系列抗氧化治疗研究中,尿酸(uric acid,UA)和米诺环素(minocycline)的基础与临床研究取得进展。UA是人体内嘌呤分解代谢的最终产物和内源性抗氧化剂[5]。在血栓栓塞性脑缺血模型,UA可抑制脑组织中性粒细胞浸润,降低ROS和活性氮水平,减轻脑梗死和神经损伤,并与溶栓药重组组织型纤溶酶原激活剂(recombinant tissue-type plasminogen activator,rtPA)产生协同作用[13-15]。临床试验结果表明,UA和rtPA联用是安全的,并可防止急性缺血性卒中(acute ischemic stroke,AIS)患者循环UA水平的早期降低,预防AIS早期缺血恶化的发生,改善功能预后[15-18]。UA还改善女性及高血糖患者的改良Rankin量表(modified Rankin scale,MRS)评分,其机制与提高女性抗氧化能力,改善葡萄糖诱导的氧化应激有关[15]。脑缺血患者入院时较高的UA水平与良好的预后相关[15]。半合成四环素-米诺环素具有抗氧化作用,可抑制iNOS表达和丙二醛(malondialdehyde,MDA)含量,上调eNOS表达,减少全脑缺血大鼠海马神经细胞死亡,提高学习记忆成绩[19-20]。随机对照临床试验发现,米诺环素能改善AIS患者MRS评分;单用米诺环素或与tPA合用有良好的耐受性和安全性,t1/2约为24 h,提示其是一种潜在的治疗AIS的神经保护药[21-22]。国家一类新药丁苯酞(nbutylphthalide,NBP)是一个多靶点抗脑缺血药物,NBP及其衍生物可明显改善缺血后线粒体能量代谢,减少细胞内ROS和MDA水平,上调内源性抗氧化系统如核因子E2相关因子2(NF-E2-related factor 2,Nrf2)、谷胱甘肽、维生素C、SOD和血红素氧合酶1等活性,减轻脑梗死和脑水肿,改善神经功能[23-26]。已上市的依达拉奉(edaravone)是一种靶向自由基的低分子抗氧化剂,具有亲脂性,易于通过细胞膜,能有效提高脑缺血后抗氧化系统活性,清除多种ROS和过氧亚硝酸盐[26-27]。一项最近的临床研究显示,AIS发作4.5 h内联合应用依达拉奉和tPA可减少症状性颅内出血及改善神经功能评分[28]。此外,国内学者还发现多种天然药物如丹参素冰片酯[29]、白杨素[30]、黄芩苷和黄芩素[31]显示有效的抗氧化损伤活性。但前期有希望的自由基清除剂NXY-059在扩大临床试验中已宣布无效[5]。
2 脑缺血后炎症反应及神经保护策略
脑缺血炎症反应在缺血性脑损伤病理过程中起重要作用。在急性期,脑组织缺血缺氧促使炎症细胞激活及小分子炎症介质如炎症细胞因子和半胱氨酰白三烯(cysteinyl leukotrienes,CysLT)上调,导致急性神经炎症和神经元损伤;在亚急性和慢性期,脑缺血炎症导致脑组织胶质细胞增生、神经元凋亡和脑组织萎缩等。这些过程均伴有神经功能的损伤。
重要炎症细胞因子白细胞介素1β(interleukin-1,IL-1β)、IL-6和肿瘤坏死因子α(tumor necrosis factor α,TNF-α)水平异常增高是导致脑缺血炎症损伤的关键因素及治疗靶标。在IL-1家族,IL-1α和IL-1β及IL-1受体拮抗剂(IL-1 receptor antagonist,IL-1Ra)与脑缺血损伤密切相关。IL-1α和IL-1β缺乏减轻小鼠脑梗死;脑缺血再灌注后IL-1β表达增高,侧脑室注射IL-1β加重缺血后脑梗死,IL-1β被认为更多参与脑缺血损伤的发病机制;而内源性IL-1Ra具有抗炎作用[5,32-33]。近年来,核苷酸结合寡聚化结构域样受体蛋白(nucleotide-binding oligomerization domain-like receptor proteins,NLRP)炎症小体与脑缺血损伤关系研究取得进展。脑缺血后,NLRP3/胱天蛋白酶1/IL-1β通路激活介导脑缺血炎症和神经元死亡,NLRP3主要表达在小胶质细胞,也表达在内皮细胞、星形胶质细胞和神经元[34-36]。除NLRP3外,NLRP1在神经元和胶质细胞表达增加与脑缺血损伤有关[34,37]。许多研究表明,脑缺血后,TNF-α和IL-6及其受体表达的增加参与脑缺血炎症和神经元死亡,抑制TNF-α和IL-6通路可减轻脑缺血损伤[32-33,38]。但也有观点认为,TNF-α和IL-6在脑缺血中具有双重作用,IL-6在急性期作为炎症介质,在亚急性和慢性期则有神经保护作用;而TNF-α和增高的TNF受体2(TNF receptor 2,TNFR2)结合与缺血预适应有关[33,39-40]。另一方面,抗炎细胞因子IL-10和转化生长因子β(transforming growth factorβ,TGF-β)被发现可限制脑缺血炎症,促进脑损伤恢复[33,41]。干预治疗研究显示,在大鼠短暂性大脑中动脉阻塞(transient middle cerebral artery occlusion,tMCAO)模型上,外源性给予IL-1Ra可减轻大鼠缺血后炎症及脑梗死,促进功能恢复[42-43],应用细胞疗法增加内源性IL-1Ra表达对持续性MCAO(permanent MCAO,pMCAO)缺血也显示有效的神经保护作用[44]。但AIS后,血浆IL-1Ra升高与高感染风险的相关性应引起注意[14]。此外,靶向炎症小体信号通路的系列措施,如NLRP IVIg、白藜芦醇、姜黄素、胱天蛋白酶1和NF-kB抑制剂等在实验研究中显示抗脑缺血作用[35-37]。研究表明,米诺环素可抑制大鼠短暂性颈总动脉阻塞(common carotid artery occlusion,CCAO)诱导的海马小胶质细胞激活及IL-1β和TNF-α水平,减轻CA1神经元死亡和记忆障碍[19]。在tMCAO和BV2细胞缺糖缺氧/恢复(oxygen and glucose deprivation/recov⁃ery,OGD/R)模型上,米诺环素通过抑制小胶质细胞NLRP3产生神经保护作用[45]。在自发性高血压大鼠,米诺环素增加tMCAO后脑灌流和紧密连接蛋白表达,降低BBB通透性,抑制小胶质细胞/巨噬细胞激活以及IL-1β和TNF-α表达,上调IL-10和TGF-β表达,进而减轻脑梗死[46];米诺环素与tPA联用不仅减轻脑缺血损伤,还可保护血管,减少tPA引起的出血,并提供更宽的治疗时间窗,提示米诺环素具有潜在的抗脑缺血应用前景[5,47]。此外,依达拉奉和NBP可抑制缺血再灌注炎症细胞因子IL-6,IL-1β和TNFα表达,但实验也发现,依达拉奉同时下调血浆IL-1Ra和IL-10水平,其机制有待进一步研究[48-49]。
近十几年来,5-脂氧合酶(5-lipoxygenase,5-LOX)/CysLT信号通路在脑缺血炎症损伤中的作用得到阐明。5-LOX产物CysLT是脑内重要的炎症介质,主要通过激动CysLT1受体(CysLT1 receptor,CysLT1R)和CysLT2R产生作用。在大鼠tMCAO模型,脑缺血中心区再灌注12~24 h,5-LOX mRNA和蛋白表达明显增高,且主要表达在神经元;在缺血周边区,再灌注3~14 d,5-LOX mRNA和蛋白表达的增高主要在增生的星形胶质细胞;脑组织5-LOX产物CysLT水平也相应增高[50]。进一步研究发现,在缺血急性期和后期,CysLT1R和CysLT2R分别在中心区神经元和小胶质细胞中以及周边区增生的星形胶质细胞中表达增加,介导脑损伤[51-52]。在体外培养细胞中,CysLT1R参与OGD/R诱导的血管内皮细胞损伤[53];CysLT2R通过细胞外信号调节激酶(extracellular signal-regu⁃lated kinase,ERK)/p38丝裂原活化蛋白激酶(p38 mitogen-activated protein kinase,p38 MAPK)通路介导星形胶质细胞水通道蛋白4表达[54]。对5-LOX抑制剂和CysLTR拮抗剂的研究表明,5-LOX非选择性抑制剂咖啡酸(caffeic acid)在体外可抑制OGD/R诱导的ROS生成和5-LOX激活,提高缺血样处理的PC12细胞存活率[55]。在短暂性全脑缺血模型上,咖啡酸抑制5-LOX表达,增加海马SOD活性,下调NF-kB表达及MDA含量,减轻缺血性脑损伤及学习记忆障碍[56];5-LOX选择性抑制剂齐留通(zileuton)也通过抑制5-LOX,下调脑组织炎症细胞因子和提高抗炎症细胞因子含量,提供神经保护作用[57]。CysLT1R拮抗剂普仑司特(pran⁃lukast)剂量依赖性地减轻大鼠和小鼠tMCAO急性期BBB破坏和神经元死亡及再灌注后14和70 d脑梗死和胶质疤痕形成,改善神经功能[58-60];另一CysLT1R拮抗剂孟鲁司特(montelukast)可预防pMCAO诱导的小鼠急性脑损伤;缺血后30 min给药仍减轻脑梗死和脑水肿;药物也减轻tMCAO小鼠和大鼠缺血后28 d脑梗死、脑萎缩及神经功能障碍[60-61]。HAMI3379是近年来报道的CysLT2R选择性拮抗剂,体外实验结果显示,其可有效抑制小胶质细胞介导的原代神经元缺血样损伤[62]。在大鼠tMCAO模型,脑室内注射HAMI3379减轻缺血后神经症状、脑梗死、脑水肿和神经元死亡,其作用与CysLT1R拮抗剂普仑司特相似[63]。HAMI3379腹腔注射可剂量和时间依赖性地抑制大鼠tMCAO诱导的小胶质细胞激活、中性粒细胞聚集、炎症细胞因子释放和脑损伤,治疗时间窗为1 h;CysLT2R基因沉默显示同样作用,提示HAMI3379的神经保护作用是CysLT2R依赖性的[64]。目前,5-LOX抑制剂和CysLT1R拮抗剂临床主要用于外周气道炎症疾病如哮喘的治疗,上述研究结果进一步提示,抗CysLT药物作为脑缺血预防和治疗药物具有潜在价值。
3 自噬在脑缺血损伤中的双重作用及自噬干预
自噬是维持细胞内环境稳态和实现自我更新的重要途径[2,65]。早在1995年,Nitatori等[66]报道短暂性CCAO可诱导沙土鼠海马CA1神经元组织蛋白酶B阳性自噬溶酶体增加和神经元损伤。近年来,自噬在缺血性神经元损伤中的作用受到关注,胶质细胞自噬与神经元损伤的关系亦被报道。但是,脑缺血后自噬的分子调控机制尚不完全清楚,自噬在脑缺血过程中起保护还是损伤作用一直存在争议[2]。
一些研究表明,脑缺血再灌注过程中自噬起神经保护作用。自噬关键调控蛋白Beclin 1作为胱天蛋白酶家族的全新底物,可被胱天蛋白酶剪切而失去活性,大鼠短暂性CCAO导致脑内凋亡蛋白胱天蛋白酶3表达增加和全长Beclin 1蛋白减少;亚精胺抑制缺血后胱天蛋白酶3活化及Beclin 1剪切,促进Beclin 1依赖的自噬,减少神经元凋亡[67]。自噬诱导剂西罗莫司(雷帕霉素,sirolimus,rapamycin)可减少新生大鼠低氧缺血诱导的脑损伤[68]。线粒体是细胞进行有氧呼吸的主要场所,脑缺血再灌注诱导线粒体损伤,线粒体自噬通过清除受损的线粒体进而发挥抗缺血性脑损伤作用。小鼠tMCAO和神经元OGD/R期间,神经元内线粒体红色荧光探针MitoTracker Red标记的线粒体与绿色荧光蛋白(green fluorescent protein,GFP)-轻链蛋白3(light chain protein 3,LC3)标记的自噬体共定位,提示线粒体自噬存在;自噬抑制剂3-甲基腺嘌呤(3-meth⁃yladenine,3-MA)和巴佛洛霉素 A1(bafilomycin A1,Baf-A1)以及自噬基因7(autophagy gene 7,Atg7)siRNA增加神经元细胞色素c释放和细胞凋亡,应用线粒体自噬抑制剂mdivi-1加重体内和体外缺血再灌注损伤,提示线粒体自噬作为脑缺血再灌注重要的内源性神经保护机制[69]。进一步研究表明,脑缺血再灌注通过诱导神经元的类NIP3样蛋白X(NIP3-like protein X,NIX)第81位丝氨酸位点的磷酸化,激活NIX介导的线粒体自噬而发挥神经保护作用,该作用是Parkin非依赖性的[70]。西罗莫司也增加tMCAO缺血线粒体Beclin 1和LC3-Ⅱ表达及p62线粒体移位,激活线粒体自噬,减轻脑梗死和神经功能障碍[71]。对缺血预处理(ischemic preconditioning,IPC)的研究发现,IPC通过适当激活自噬产生缺血耐受作用,减轻pMCAO诱导的脑梗死和运动障碍;3-MA和Baf-A1对抗IPC的神经保护作用,西罗莫司可模仿IPC的作用[72]。原代皮质细胞OGD实验发现,IPC或西罗莫司的作用与抑制过度内质网应激有关[73]。酸后处理作为内源性保护策略,通过增强Parkin依赖的线粒体自噬,对体内、外缺血神经元有明显保护作用,并延长脑缺血治疗时间窗[74]。
另一方面研究表明,长期和持久的脑缺血触发神经细胞过度自噬可诱导自噬性细胞死亡,进而加重缺血性脑损伤[2,75]。在大鼠pMCAO结合短暂性CCAO模型,再灌注后立即开展缺血后处理(isch⁃emic postconditioning,IPOC)可抑制缺血诱导的LC3/Beclin 1表达和p62下降,减轻脑梗死和脑水肿;西罗莫司部分逆转IPOC的作用,而3-MA通过抑制缺血后自噬激活以及促进抗凋亡蛋白Bcl-2表达,产生与IPOC一致的神经保护作用[76]。在原代皮质神经元或SH-SY5Y细胞培养,6 h OGD/R诱导神经元自噬性死亡,3-MA抑制OGD/R诱导的神经元死亡[77]。在小鼠pMCAO模型,缺血缺氧诱导小胶质细胞自噬与脑损伤,3-MA抑制细胞自噬显著减少脑梗死、脑水肿和神经功能缺损,提示过度的小胶质细胞自噬参与了脑缺血损伤过程[78]。随着对自噬在脑缺血损伤中调节机制研究的不断深入,自噬可能成为脑缺血治疗的新靶点。
4 脑缺血损伤中氧化应激、炎症和自噬的相互作用
新的证据表明,脑缺血损伤中氧化应激、炎症与自噬存在着紧密和复杂的相互作用。脑缺血损伤过程中氧化应激、炎症和自噬的相互作用及潜在的治疗干预措施总结见图1。①脑缺血再灌注后氧化应激调控细胞自噬。脑缺血及再灌注后迅速增加的ROS诱导自噬的核内相关信号通路包括:ROS-p53-糖酵解和凋亡调节因子(glycolysis and apoptosis regulator,TIGAR)/损伤调控自噬调节因子(damage-regulated autophagy modulator,DRAM)通路;ROS-低氧诱导因子1(hypoxia-inducible factor 1,HIF1)-Bcl-2/腺病毒E1B相互作用蛋白3(Bcl-2/adenovirus E1B interacting protein 3,BNIP3)和NIX通路;ROS-Nrf2-p62以及ROS-叉头框蛋白O3(forkhead box O3,FoxO3)-LC3/BNIP3通路;此外,ROS也调节蛋白激酶R样内质网激酶(protein kinase R-like ER kinase,PERK)及其下游相关基因诱导自噬[2,79]。在细胞浆,ROS通过抑制自噬体至自噬溶酶体形成过程中Atg4活性进而诱导自噬[79]。线粒体自噬是神经细胞对线粒体实行数量和质量控制的重要手段。脑缺血再灌注后过度增加的ROS可触发细胞内钙超载,进而导致线粒体功能障碍和线粒体自噬,线粒体自噬可被Parkin依赖和非依赖性途径所介导[2,70,79-80]。但脑缺血损伤中NOS/NO对自噬的调节作用尚未完全阐明。在大鼠原代皮质神经元,NO可通过促进JNK1和IKKβ的亚硝基化抑制其生物活性,进而抑制细胞自噬[81]。②脑缺血后氧化应激促进炎症反应。脑缺血再灌注后过量的ROS产物可诱导NF-κB转录因子活化,进一步促进炎症分子包括TNF-α、IL-6和NLRP3/胱天蛋白酶1/IL-1β上调,导致脑缺血后炎症和神经损伤[2,5,82]。抗氧化剂依达拉奉通过保护线粒体正常结构和减少ROS水平,进而抑制体内、外缺血诱导的5-LOX/CysLT通路激活,产生神经保护作用[83]。③自噬负调节脑缺血后氧化应激反应。脑缺血再灌注后适度激活的线粒体自噬通过自噬性清除缺血损伤的线粒体,阻断ROS与线粒体相互作用形成的“ROS诱导的ROS释放”(ROS-induced ROS release),对缺血后神经元生存有重要意义[2-3]。④自噬调节缺血后神经炎症。自噬诱导剂西罗莫司通过抑制mTOR通路,促进自噬相关蛋白Beclin 1和LC3Ⅱ表达,预防新生小鼠缺氧缺血诱导的小胶质细胞激活和神经元死亡[84]。白藜芦醇诱导去乙酰化酶沉默信息调节因子1依赖的细胞自噬,可抑制NLRP3激活,减轻脑缺血再灌注损伤[85]。但也有报道小胶质细胞自噬参与脑缺血后神经炎症[78]。⑤细胞炎症诱导自噬激活。在巨噬细胞过表达NLRP3/IL-1β诱导的自噬激活,可促进NLRP3降解及抑制下游炎症产物胱天蛋白酶1和IL-1β水平;3-MA抑制自噬增强NLRP3/胱天蛋白酶1/IL-1β活性,而西罗莫司诱导自噬抑制其活性[86-88]。
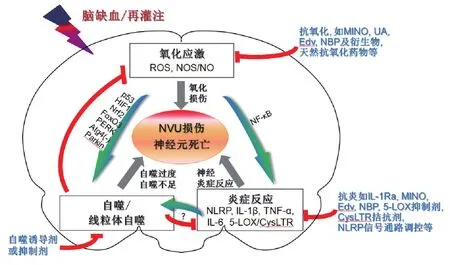
图1 脑缺血再灌注过程中,氧化应激、炎症和自噬相互作用及潜在治疗干预措施.Atg4:自噬基因4;CysLTR:半胱氨酰白三烯受体;Edv:依达拉奉;FoxO3:叉头框蛋白O3;HIF1:缺氧诱导因子1;IL-1β:白细胞介素1β;IL-1Ra:IL-1受体拮抗剂;IL-6:白细胞介素6;5-LOX:5-脂氧合酶;MINO:米诺环素;NBP:丁苯酞;NF-κB:核因子-κB;NLRP:核苷酸结合寡聚化结构域样受体蛋白;NO:一氧化氮;NOS:一氧化氮合酶;Nrf2:核因子E2相关因子2;NVU:神经血管单元;PERK:蛋白激酶R样内质网激酶;ROS:活性氧;TNF-α:肿瘤坏死因子α;UA:尿酸.
5 结语
氧化应激和神经炎症在脑缺血损伤过程中的重要作用已得到共识。自噬作为一种新的脑缺血调节机制正得到越来越多的关注。最新研究表明,脑缺血再灌注病理过程中氧化应激、炎症和自噬相互作用,构成一个复杂的信号网络,在脑缺血级联事件中起关键作用。因此,单一针对某一机制的药物治疗可能不足以对抗缺血再灌注导致的脑损伤,开展多靶点多环节的神经保护药物研究将是抗脑缺血研究的发展趋势。近年来,一些药物如UA[13,15-18]、米诺环素[19-22,45-47]、依达拉奉[26-28,48,83]、NBP及其衍生物[23-26,49]以及5-LOX/CysLT信号通路抑制剂[55-64]在脑缺血基础研究中显示抗氧化抗炎等多重神经保护作用。此外,为避免目前使用的自噬诱导剂和抑制剂以及自噬基因敲除对神经细胞正常功能的影响,一些研究已专注于调控脑缺血后自噬活性的新化合物研究[67,85,89-90]。深入揭示氧化应激、炎症和自噬对脑缺血再灌注损伤的调控作用及潜在治疗靶点,对于正确理解缺血性脑损伤的病理生理机制,以及推动新的、有效的神经保护药物研发具有重要的理论和实际意义。
猜你喜欢
医学研究生学报(2022年5期)2022-12-07
中华实用诊断与治疗杂志(2022年1期)2022-08-31
昆明医科大学学报(2021年8期)2021-08-13
海洋通报(2021年1期)2021-07-23
昆明医科大学学报(2021年3期)2021-07-22
天津医科大学学报(2021年3期)2021-07-21
生物学通报(2021年4期)2021-03-16
昆明医科大学学报(2020年11期)2020-12-28
神经损伤与功能重建(2020年10期)2020-12-23
神经损伤与功能重建(2020年11期)2020-12-01
