
植物乳杆菌降胆固醇Lp10菌株的rhaD基因敲除株的构建
2018-12-22 07:14:50郭天芬林俊芳郭丽琼叶志伟魏韬许之恒
现代食品科技 2018年11期
郭天芬,林俊芳,2,郭丽琼,2,叶志伟,2,魏韬,2,许之恒
(1.华南农业大学食品学院生物工程系,食品生物技术研究所,广东广州 510642)
(2.广东省微生态制剂工程技术研究中心,广东广州 510642)
血清中高浓度的胆固醇是诱发高血压、冠心病等众多心血管疾病的重要因素[1],控制血清胆固醇水平已经受到越来越广泛的关注。传统药物治疗存在药费昂贵,毒副作用较大、治疗效果不明显等缺陷[2]。Mathara等[3]从传统发酵乳中分离出了23株乳酸菌,分析显示部分乳杆菌具有降胆固醇的能力。益生菌对降胆固醇的治疗效果明显,对高血脂的防治有积极作用,并有望替代药物治疗法成为了更安全、更有效的降胆固醇方法[4,5]。
植物乳杆菌(Lactobacillus plantarum)是具有降胆固醇[6]、改善免疫功能[7],维持肠道菌群平衡[8],抑制肿瘤细胞的形成[9]等益生功能的革兰氏阳性细菌。目前,植物乳杆菌降胆固醇的作用机理主要有以下几种观点:(1)细胞直接吸收胆固醇;(2)胆盐水解酶(bile salt hydrolase,BSH)作用;(3)胞外多糖(exopolysaccharide,EPS)的吸附作用;(4)鉴于植物乳杆菌降低胆固醇效果受多种因素的影响,还存在一些其他理论[10]。
植物乳杆菌的次级代谢产物EPS种类繁多,其组分包括果糖、葡萄糖、甘露糖、半乳糖和鼠李糖等。L-鼠李树胶糖-1-磷酸醛缩酶(L-rhamnulose-1-phosphate aldolase,RhaD)按照供体专一性进行分类,属于磷酸二羟基丙酮(DHAP)依赖性醛缩酶,在植物乳杆菌中参与L-鼠李糖的代谢。首先,L-鼠李糖在L-鼠李糖异构酶的催化下生成L-鼠李树胶糖,然后经过L-鼠李树胶糖激酶的作用生成L-鼠李树胶糖-1-磷酸,最后经RhaD裂解为L-乳醛和磷酸甘油酮,进入糖酵解循环[11,12]。研究组前期的转录组分析结果表明,在植物乳杆菌Lp10菌株降胆固醇过程中rhaD基因表现为转录水平下调,推测植物乳杆菌在添加胆固醇环境下主要是通过产生EPS对胆固醇进行吸附,而此过程中植物乳杆菌为提高EPS的合成量以下调rhaD基因的表达量来抑制了L-鼠李糖的代谢。为了深入分析rhaD基因与植物乳杆菌Lp10菌株降胆固醇功能的关系,本研究利用同源重组原理敲除rhaD基因,并比较初始菌株与突变菌株的降胆固醇能力。同源重组技术在乳酸菌中已经应用广泛,例如李娅妮等[13]在pUC18敲除载体的基础上成功构建了基因敲除载体pUC18△lai并实现对植物乳杆菌的亚油酸异构酶(Linoleic isomerase,lai)基因的敲除;岳云春等[14]利用同源重组法构建质粒pKLKRT,并电转化于副干酪乳杆菌(Lactobacillus paracasei)感受态细胞,成功构建组氨酸蛋白激酶基因缺失突变株。
植物乳杆菌Lp10菌株是本研究组前期从海带中分离出来的一株具有高降胆固醇能力的乳酸菌[15]。本实验选用pNZ5319作为敲除质粒,构建rhaD基因敲除载体,试图摸索出一套完整的植物乳杆菌基因敲除载体的构建及转化体系,为植物乳杆菌降胆固醇机制及其功能基因的分析研究奠定实验基础。
1 材料与方法
1.1 材料
1.1.1 菌株与质粒
植物乳杆菌(Lactobacillus plantarum)Lp10菌株由本研究组从海带中分离纯化并保存;大肠杆菌(Escherichia coli)DH5α购买于大连TaKaRa公司;质粒pNZ5319由南开大学乔明强教授惠赠。
1.1.2 主要试剂和培养基
细菌基因组DNA提取试剂盒购买于TIANGEN公司;琼脂糖凝胶DNA纯化回收试剂盒、质粒小提试剂盒购买于MAGEN公司;Ex Taq酶购买于大连Takara公司;限制性内切酶XhoI、PmeI、SacI、BglII购买于 Thermo Fisher Scientific公司;氯霉素(chloramphenicol,cm)购于Solarbio公司;其余试剂均为分析纯。
MRS培养基(1 L):蛋白胨10 g、葡萄糖20 g、牛肉膏10 g、酵母粉5 g、磷酸氢二钾2 g、乙酸钠5 g、柠檬酸氢二铵2 g、吐温-80 1 mL、硫酸镁0.58 g、硫酸锰0.25 g、pH 6.8、琼脂20 g(固体培养基添加)。
LB培养基(1 L):胰蛋白胨10 g、酵母提取物5 g、NaCl 10 g、pH 7.0、琼脂粉 20 g(固体培养基添加)。
恢复培养基:含0.3 mol/L蔗糖、20 mmol/L MgCl2、2 mmol/L CaCl2的MRS培养基。
1.1.3 仪器
SPX-158L生化培养箱宁波新芝生物科技股份有限公司;FRESC21微量台式冷冻离心机Thermo Fisher Scientific公司;YXQ50蒸汽灭菌锅西安太康生物科技公司;BIO-RADC1000梯度PCR仪和Gel DocTMXR+凝胶成像系统BIO-RAD公司;SKJH-1109超净工作台上海苏坤实业公司;EPS-300电泳仪上海天能公司。
1.1.4 引物
本文所用引物如表1所示。
1.2 实验方法
1.2.1rhaD基因同源臂的克隆及纯化
根据GenBank中已登录的植物乳杆菌rhaD基因两侧序列,设计引物rhaD-up-F/rhaD-up-R、rhaD-down-F/rhaD-down-R分别扩增长约为 1200 bp的rhaD基因上游同源臂(rhaD-up)和下游同源臂(rhaD-down),并在rhaD-up片段引入限制性酶切位点XhoI和PmeI,rhaD-down片段引入限制性酶切位点SacI、BglII。植物乳杆菌Lp10菌株基因组DNA按照TIANGEN细菌基因组DNA提取试剂盒说明书步骤提取。PCR反应体系(50 μL):模板DNA(225 ng/μL)2 μL,10×Ex Taq buffer(20 mM)5 μL,dNTPs(2.5 mM)4 μL,上下游引物(20 μM)各1 μL,Ex Taq 酶(5 U/μL)0.5 μL,ddH2O 补足 50 μL。PCR 条件为:94 ℃预变性 5 min;94 ℃变性 30 s,65 ℃(rhaD-up)或68 ℃(rhaD-down)退火30 s,72 ℃延伸72 s,34个循环;72 ℃延伸10 min,4 ℃保存。PCR产物经1%琼脂糖凝胶电泳后,切下目的条带并使用琼脂糖凝胶DNA纯化回收试剂盒回收目的片段并进行测序比对分析。
1.2.2rhaD基因敲除质粒的构建
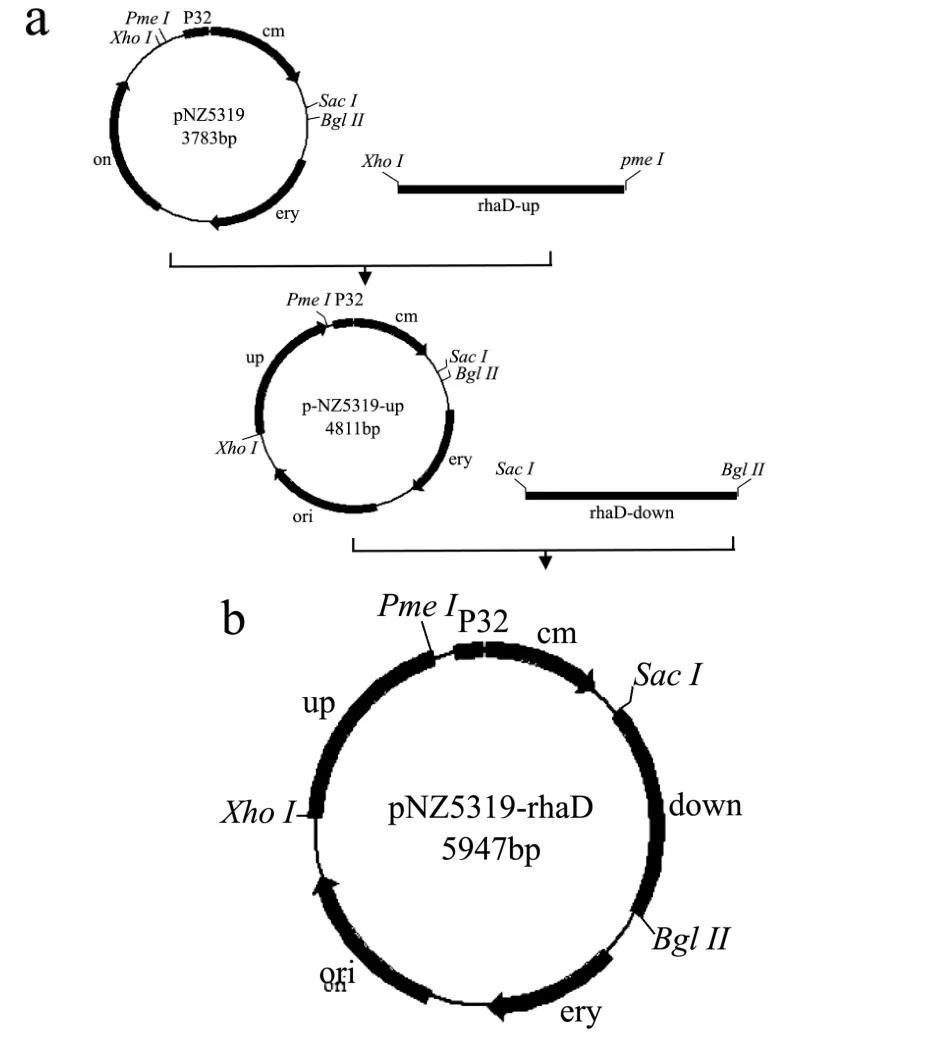
图1 敲除载体pNZ5319-rhaD构建路线Fig.1 Structure route of knockout vector pNZ5319-rhaD
植物乳杆菌rhaD基因的pNZ5319-rhaD敲除质粒的构建路线如图1所示。利用限制性内切酶XhoI和PmeI分别对pNZ5319质粒和rhaD-up进行双酶切并进行琼脂糖电泳回收,利用abm必克隆试剂盒将双酶切处理后的pNZ5319质粒和rhaD-up片段进行连接处理,转化DH5α感受态细胞并于终浓度为15 μL/mL氯霉素的LB固体培养基过夜培养。
挑取转化子并扩大培养后,提取质粒进行XhoI和PmeI双酶切鉴定和测序鉴定,将正确的重组质粒命名为pNZ5319-up。
分别将 pNZ5319-up质粒和下游同源臂rhaD-down片段进行SacI和BglII双酶切和回收处理。将双酶切后的pNZ5319-up质粒和rhaD-down片段连接并转化E. coliDH5α感受态细胞。对重组质粒进行酶切鉴定和测序鉴定(参照1.2.2.1),将正确的重组质粒命名为pNZ5319-rhaD。
1.2.3 植物乳杆菌感受态细胞的制备
植物乳杆菌 Lp10菌株按 1%接种量接种于 100 mL含1%甘氨酸的MRS培养基,37 ℃,静置培养至OD600约0.8(6~7 h);冰浴15 min,使菌停止生长;于4 ℃,8000 g下离心5 min后收集菌体;用100 mL冰浴的30% PEG4000洗涤3次,离心条件同上;用1 mL冰浴的30% PEG4000重悬菌体,分装每管100 μL,-80 ℃保存。
1.2.4 电转化
将20 μL 重组质粒(浓度537.8 ng/μL)与100 μL植物乳杆菌 Lp 10菌株感受态细胞在冰浴条件下混匀,转移至电转杯中,冰浴10 min。在电阻200 Ω,电容值25 μF,电压2.5 kV条件下进行电击,电击后迅速加入800 μL恢复培养基,混合均匀后37 ℃静置培养3 h并涂布于含终浓度为5 μg/mg氯霉素的MRS培养基,于37 ℃培养24~48 h。
1.2.5 植物乳杆菌Lp10rhaD基因敲除突变株的鉴定
利用TIANGEN细菌基因组DNA提取试剂盒(离心柱)提取植物乳杆菌 Lp10初始菌株和重组转化子的基因组 DNA。以重组转化子和初始菌株基因组DNA为模板,分别扩增上游同源臂rhaD-up、下游同源臂rhaD-down、cm基因和rhaD基因等DNA片段。
1.2.6 植物乳杆菌Lp10rhaD基因敲除突变株的生长分析
将植物乳杆菌突变株与初始菌株按接种量 1%接种于MRS液体培养基中,37 ℃静置培养,以0 h为起点,每隔2 h取一次样并测定OD600,测定到24 h,通过软件Origin绘制生长曲线,观察两株菌的生长情况。
1.2.7 植物乳杆菌Lp10rhaD基因敲除突变株的降胆固醇能力测定
将植物乳杆菌突变株与初始菌株分别接种于加有1%胆固醇溶液的MRS培养基中,37 ℃培养20 h。
按照邻苯二甲醛法[15]对初始菌株和突变菌株进行降胆固醇能力的测定,每个样品进行3次重复进行试验。
1.2.8 数据统计分析
实验中每个数据重复三次,结果表示为平均数±标准差。采用Origin 8.5软件作图,SPSS 17.0进行显著性分析。
2 结果与分析
2.1rhaD基因同源臂的克隆及纯化
以植物乳杆菌Lp10菌株基因组DNA为模板,分别扩增rhaD基因上下游同源臂,将PCR产物进行1%琼脂糖凝胶电泳,发现在1200 bp处都有明显的单一条带,与预计大小一致(图2)。目的条带的测序鉴定表明rhaD基因上下游同源臂被成功克隆。

图2 rhaD基因同源臂的PCR扩增Fig.2 PCR amplification for homologous arms of rhaD gene
2.2 同源臂与pNZ5319载体的连接及转化
对上游同源臂rhaD-up片段与敲除载体pNZ5319连接所得的pNZ5319-up转化子进行菌液PCR鉴定及XhoI和PmeI双酶切鉴定。PCR凝胶电泳结果显示在1200 bp处有一清晰条带,与目的片段rhaD-up大小相符(图3a);双酶切pNZ5319-up质粒,得到2个片段,大小与pNZ5319质粒和rhaD-up相符(图3b)。同理,得到的 pNZ5319-rhaD转化子进行菌液 PCR鉴定及SacI和BglII双酶切鉴定(图3c和3d)。DNA测序结果进一步验证敲除载体 pNZ5319-rhaD被成功构建。

图3 敲除载体转化子的鉴定Fig.3 Identification of transformants with knockout vector
2.3 重组转化子的鉴定
利用引物rhaD-up/rhaD-down、cm-up/cm-down对植物乳杆菌初始菌株和重组转化株进行PCR鉴定。实验结果显示,从植物乳杆菌 Lp10初始菌株基因组DNA中能扩增到与目的基因rhaD(852 bp)大小相同的片段,而不能从重组转化株的基因组 DNA和ddH2O(阴性对照)中扩增出相同的片段(图 4b);从重组转化株基因组DNA中能扩增出与目的基因cm(651 bp)大小相同的片段,而不能从初始菌株基因组DNA和ddH2O中扩增出相同片段(图4c)。分别将以上扩增所得的两个片段进行回收和测序鉴定,表明两者分别为rhaD和cm基因。因此,重组转化株的rhaD基因被成功敲除。

图4 植物乳杆菌Lp10重组转化株的鉴定Fig.4 Identification of recombinant transformation of L.plantarumLp10
2.4 植物乳杆菌Lp10rhaD基因敲除突变株的生长曲线

图5 突变菌株和初始菌株的生长曲线Fig.5 growth curve of mutant strain and original strain
植物乳杆菌Lp10rhaD基因敲除突变株与初始菌株在MRS培养基中的生长曲线如图5,两株菌都符合典型的生长曲线趋势,在4 h进入对数生长期,14 h后进入稳定生长期,但突变株进入稳定生长期后相比于初始菌株生长稍缓慢。
2.5 植物乳杆菌Lp10rhaD基因敲除突变株的降胆固醇能力
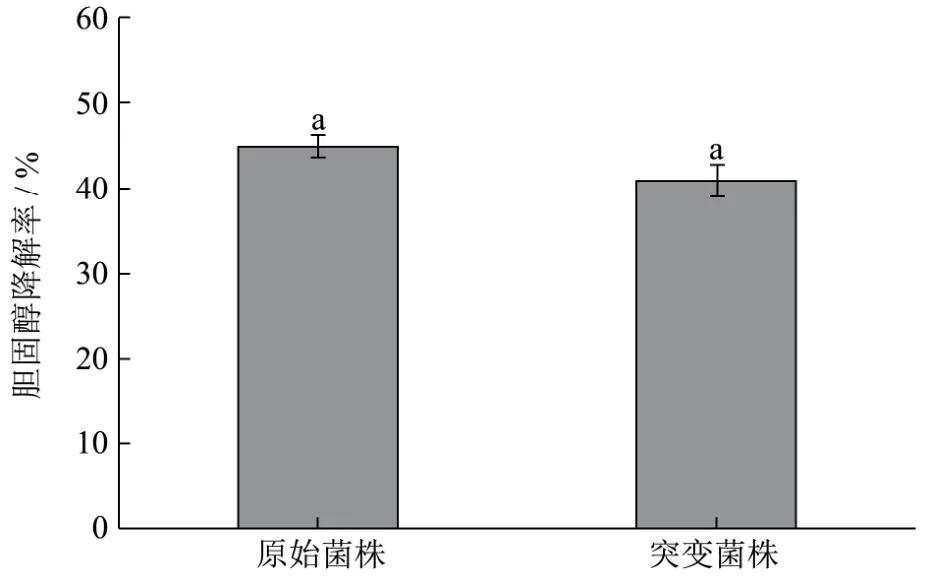
图6 胆固醇能力的测定Fig.6 Determination of cholesterol lowering ability
根据邻苯二甲醛法测定胆固醇降解率,据图6可知,初始菌株的胆固醇降解率为45%,突变菌株的胆固醇降解率为41%,两株菌株的 OD600分别为2.373和 2.133,即初始菌株的胆固醇降解率为18.96%/OD600,敲除菌株的胆固醇降解率为19.22%/OD600,结果表明基因rhaD敲除后,植物乳杆菌 Lp10的降胆固醇能力略有上升,但差异不显著(p>0.05)。
3 结论
3.1 近年来,利用组学研究挖掘差异基因已经成为趋势,例如基因组、转录组以及蛋白组学等,了解差异基因的功能已成为研究的热点和核心内容。研究基因功能的一个重要方法,就是对该基因进行敲除,获得突变株,然后根据突变株的变化来推测基因产物的功能[17]。本研究基于植物乳杆菌Lp10降胆固醇过程的转录组数据,通过GO数据库和KEGG代谢通路数据分析,选择rhaD基因为研究对象。利用同源重组原理,成功获得植物乳杆菌Lp10rhaD基因敲除突变株。对初始菌株与敲除菌株进行生长曲线及降胆固醇能力测定,结果表明,敲除菌株的生长较初始菌株更缓慢,初始菌株的胆固醇降解率为18.96%/OD600,敲除菌株的胆固醇降解率为19.22%/OD600,表明基因rhaD基因经敲除后,植物乳杆菌 Lp10的降胆固醇能力略有上升,但差异不显著,推测可能原因是敲除载体pNZ5319-rhaD在植物乳杆菌Lp10发生同源重组时,cm抗性基因与rhaD基因发生互换,cm基因整合到基因组上,对植物乳杆菌 Lp10生长、生理活动等产生负影响,后续研究可将Cre-loxP系统[14]转化至突变株中,对抗性筛选标记cm基因进行敲除,此外,rhaD基因的敲除对菌体生长也有所影响,该基因参与鼠李糖在生物体内的代谢,且鼠李糖属于细胞初级代谢产物,rhaD基因的敲除影响菌体细胞正常的初级代谢,从而导致菌体生长减缓,菌体量下降。
3.2 本研究构建pNZ5319-rhaD敲除载体时,考虑同源重组效率与同源臂长度有一定的关系,在0.3~1.2 kb范围内,随着同源臂长度的增加,同源重组效率呈对数增加[14],rhaD基因的上下游同源臂rhaD-up/rhaD-down长度都约为1200 bp,有利于目标基因与抗性筛选基因发生同源重组。本研究选用的载体pNZ5319是植物乳杆菌-大肠杆菌穿梭载体,氯霉素抗性筛选标记在植物乳杆菌和大肠杆菌中都能发挥作用,无需其他抗性基因的引入,简化了实验操作。
3.3 近年来,随着分子生物学的大力发展,基因敲除技术成为了热点。同源重组技术已成功应用于乳酸菌中,但存在效率低、工作量大等缺点,通过合理设计同源臂、选择合适的敲除载体、优化电击条件均可以提高同源重组效率。除了传统的同源重组技术,CRISPR/Cas技术已日渐成熟[18,19]。Jiang等[20]利用CRISPR/Cas系统成功实现了原核生物基因的定向修饰。后续研究可尝试利用CRISPR/Cas技术建立和完善的植物乳杆菌基因操作系统。
3.4 植物乳杆菌敲除系统可以用于降胆固醇功能性植物乳杆菌的开发,应用于酸奶、乳酸菌饮料及益生菌制剂等方面达到降胆固醇的目的,弥补药物治疗引发的肝肾受损、戒断反应和复发等副作用,减少心脑血管发病率,并且食品级敲除菌株包括独特风味,成本低,效率高等优点,具有一定的市场前景。
3.5 本研究成功建立了一套完整的植物乳杆菌敲除体系及电转化体系,这不仅为植物乳杆菌基因敲除载体的构建提供了一定的实验经验,也为后续 rhaD基因的功能验证奠定了基础。为了进一步深入验证rhaD基因与EPS吸附作用降胆固醇机制的关系,后续研究将对敲除前后菌体表面的EPS含量及种类进行分析鉴定,为高效降胆固醇乳酸菌制剂的研究和开发利用提供实验基础和理论依据。
猜你喜欢
——紫 苏
河南农业(2024年1期)2024-01-19 01:56:54
华人时刊(2023年1期)2023-03-14 06:43:36
保健医苑(2022年1期)2022-08-30 08:39:52
中老年保健(2022年3期)2022-08-24 02:58:40
中老年保健(2022年4期)2022-08-22 02:59:58
汉字汉语研究(2021年2期)2021-08-30 08:58:46
今日农业(2021年11期)2021-08-13 08:53:24
饮食科学(2017年12期)2018-01-02 09:23:27
河北书画研究(2016年3期)2016-04-28 08:55:35
遗传(2014年3期)2014-02-28 20:58:49
