
外场作用下C12H4Cl4O2的分子结构和电子光谱研究∗
2018-12-18 05:57杜建宾冯志芳韩丽君唐延林武德起
物理学报 2018年22期
杜建宾 冯志芳 韩丽君 唐延林 武德起
1)(廊坊师范学院物理与电子信息学院,廊坊 065000)
2)(廊坊师范学院数学与信息科学学院,廊坊 065000)
3)(贵州大学物理学院,贵阳 550025)
4)(河南机电职业学院信息工程学院,郑州 451191)
(2018年7月30日收到;2018年9月12日收到修改稿)
1 引 言
多氯代二苯并-对-二噁英(PCDDs)、多氯代二苯并呋喃(PCDFs)和类二噁英多氯联苯(dl-PCBs)因具有相似的结构和毒性特征,统称为二噁英类[1].它们不仅具有很强的毒性,而且具有生物富集性、致突变性以及在环境介质中的稳定性[2],这对人类健康和生态环境具有严重的危害[3].二噁英类物质中以C12H4Cl4O2(2,3,7,8-tetrachlorodibenzo-p-dioxin,TCDD)毒性最强,是目前已知毒性最强的化合物[4].Miyazaki等[5]研究了TCDD对血脑障壁的影响,证实了婴儿中枢神经对其有不良反应.Fracchiolla等[6]证实了TCDD可引起各种血液疾病.杜国勇等[7]对垃圾焚烧厂区二噁英污染及厂区工人呼吸暴露问题进行了评估,证实厂区空气质量处于毒性较高水平.因此,去除TCDD成为近年来讨论的热点问题.生物光解TCDD[4]、利用黑磷和掺杂Ni的氮化硼纳米管等新兴材料吸附TCDD等方法已被采用[8,9].
在外电场的作用下,有机物会出现一些新的现象,比如新自由基的产生[10−13]、化学键碎裂、新激发态出现以及振动斯塔克效应[14]等[11,12,15−17].吴永刚等[18]对外电场作用下的CdSe分子进行了研究,结果表明分子结构受外电场影响变化明显;谢安东等[19]对UO3分子在自辐射场下的光谱进行了计算,结果显示分子能级对自辐射场有强依赖性;王藩侯等[10]发现外电场对SnSe的能级和光谱等有明显影响.但在外电场作用下,TCDD分子结构和光谱的理论计算目前还未见报道.本文首先采用密度泛函理论(density functional theory,DFT)[8,9]B3LYP[20,21]方法,在6-31+g(d,p)[20]基组水平上对TCDD分子的基态几何结构和总能量在不同电场下的变化进行了研究,然后在同样基组的水平上采用含时密度泛函理论(time-dependent density functional theory,TDDFT)[18,22−24]方法研究了外电场对TCDD分子紫外-可见(UV-Vis)吸收光谱产生的影响,这为TCDD的检测和降解方法研究提供了理论依据.
2 理论与计算方法
在外电场的作用下,分子体系的哈密顿量H[25]由两项组成:

其中H0为无电场时的哈密顿量,Hint为分子体系与外电场相互作用的哈密顿量.Hint在偶极近似时表示为

其中µ为分子体系的电偶极矩,F为外电场的强度.
忽略外电场下极化率的非线性效应,由文献[23,26]提出的模型可得,激发能Eexc与极化率的变化量∆α、电偶极矩变化量∆µ和电场强度F满足的关系式为

其中Eexc(0)为无电场时的激发能.振子强度f1u为[25,27]

其中gl为加权因子,这里等于1;σ表示能量,单位为cm−1;线强度S为原子单位(e2a20).本文在Grimme的半经验方法基础上,把和电场相关的项加入到哈密顿量中[19,28],并采用TDDFT[20]来精确计算激发能.
TCDD分子的结构如图1所示,本文在沿x轴方向上加上一系列强度为0—0.025 a.u.(0—1.2856×1010V/m)的外电场(限于篇幅,本文只讨论沿x轴方向加外电场的情况,其他方向以后另行讨论),也就是把Hint添加到Gaussian程序的哈密顿量中,然后采用DFT/B3LYP/6-31+g(d,p)方法,优化了TCDD分子在不同外电场作用下的基态几何结构,其收敛标准如表1所列,同时得到了分子总能量,最后采用TDDFT方法研究了外电场对分子前26个激发态的波长和摩尔吸收系数产生的影响.全部计算在Gaussian09软件包中完成.
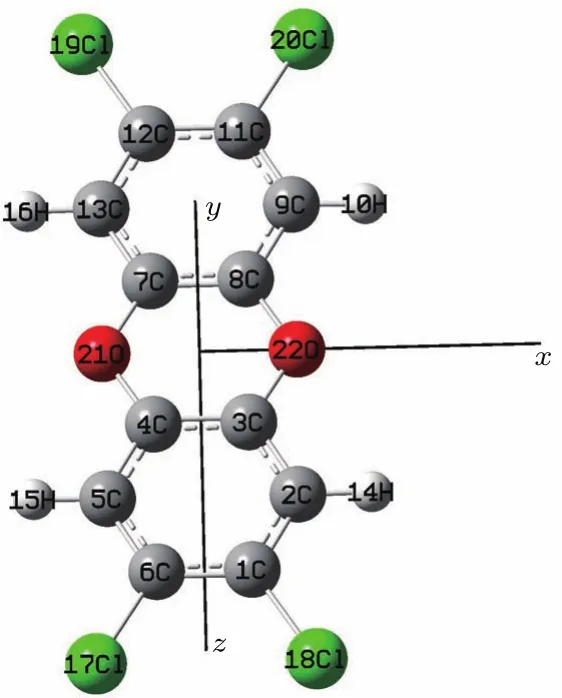
图1 优化的TCDD分子结构Fig.1.Optimized structure of the TCDD molecule.

表1 Gaussian09软件收敛标准Table 1.The convergent standard of Gaussian09 software.
3 不同外电场下TCDD分子的几何结构
采用DFT方法优化不同外电场作用下TCDD分子的基态几何结构,得到的几何参数、密立根电荷和分子总能量分别列在表2—表4中,限于篇幅,我们只选择了主要的键长和部分原子的密立根电荷予以列出.随着外电场的增加,TCDD分子几何参数变化明显,如表2及图2所示.在0—0.025 a.u.范围内的外电场作用下,R(2,14),R(12,19)和R(4,21)等随着外电场的增强先减小后增加,而R(11,20)和R(3,22)等随着外电场的增强先增加后减小,这些变化可以用分子内电场与外电场的叠加效应来解释[23].当外电场较弱时,电子的局域偏移使得2C-14H,12C-19Cl和4C-21O之间的电场增强,从而其键长减小.2C-14H中的C,12C-19Cl中的Cl和4C-21O中的O电负性较强,在较弱的外电场的作用下,原本偏向C,Cl和O的电子云进一步向其偏移,使得两原子间电场增强,键长减小:2C在无外电场时,密立根电荷为−0.087057 a.u.,当外电场增加到0.005 a.u.时,局部电子云的偏移使得2C的密立根电荷增加到−0.131662 a.u.,如表3所列.但随着外电场的增强,电子发生了整体偏移,外电场与2C-14H,12C-19Cl和4C-21O原子间的内电场方向相反,从而使得原子间的叠加电场减弱,键长增加:电子的整体偏移使得2C带上了正电荷,随着外电场的增强,2C和14H之间的作用力越来越弱,从而键长增加,如表2和表3所列.与之相反,当外电场较弱时,电子的局域偏移使得11C-20Cl和3C-22O之间的电场减弱,从而其键长增加;但随着外电场的增强,电子发生了整体偏移,外电场与11C-20Cl和3C-22O原子间的内电场方向相同,从而使得原子间的叠加电场增强,键长减小.

表2 不同电场下TCDD分子的基态键长R/ÅTable 2.Optimized bond lengths(unit:Å)for the ground states of the TCDD molecule under different external electric fields.

表3 不同电场下原子2C和14H的电荷Q/a.u.Table 3.The charges of 2C and 14H(unit:a.u.)of the TCDD molecule under different external electric fields.

表4 基态总能量E/a.u.随外电场F/a.u.变化的关系Table 4.Variation of total energies(unit:a.u.)of TCDD molecule with electric field intensities(unit:a.u.).

图2 TCDD分子主要键长R随外电场变化Fig.2.Relationship between the main bond lengths of TCDD molecule and electric field intensities.
TCDD分子的基态总能量随着外电场的增强而减小,如表4和图3所示,F=0 a.u.时,基态总能量E=−2450.921034 a.u.,但当F=0.025 a.u.时,基态总能量E减小到了−2450.983302 a.u.,且减小的趋势加剧,这是由于外电场与内电场的叠加使得分子几何结构发生了变化,如图2所示,无外电场时,分子结构松动,基态总能量较大,但随着外电场的增强,分子结构变得更加稳固,基态总能量也急剧减小,计算结果与(1)式也是一致的.分子几何结构的变化,也使得分子点群从C2h变为CS.

图3 分子总能量E随外电场的变化Fig.3. Relationship between the total energies of TCDD molecule and electric field intensities.
4 TCDD分子的激发态波长以及外电场对激发态波长和摩尔吸收系数的影响
4.1 TCDD的激发态波长
在分子基态几何结构优化的基础上,采用TDDFT方法计算了TCDD分子的前26个激发态,得到了分子的UV-Vis吸收光谱,如图4所示,有波长为221 nm、摩尔吸收系数为54064 L·mol−1·cm−1的强吸收峰,处于E带,它是环状共轭的三个乙烯键的苯型体系中的π→ π∗电子跃迁产生的;此外,在波长为296 nm处有一摩尔吸收系数为6234 L·mol−1·cm−1的肩峰, 它是芳香族化合物π→π∗电子跃迁的特征吸收,处于B带.Koshiok等[29]利用Shimadzu UV-240光谱仪,测量了以1,4-二氧六环为溶剂、浓度为50 ppm的TCDD溶液在260—400 nm波长范围的紫外光谱,结果显示在305 nm处有一个强吸收峰.与文献中的数据对比,计算的肩峰波长仅有9 nm的蓝移,考虑到实验误差、溶剂效应等原因,可以看出,我们的计算精确度是可信的,并且我们的计算是对文献很好的补充.

图4 TCDD分子在不同外电场作用下的UV-Vis吸收光谱Fig.4.UV-vis absorption spectra of TCDD molecule under different external electric fields.
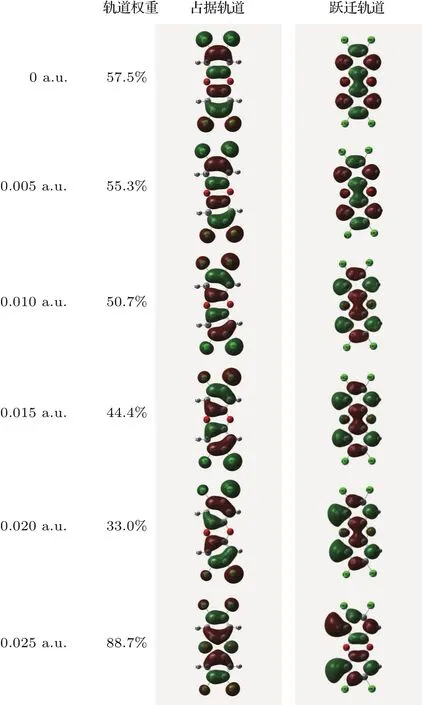
图5 不同外电场下TCDD激发态的分子前线轨道Fig.5.Excited state frontier orbital diagram of TCDD molecule under different external electric fields.
4.2 外电场对激发态波长和摩尔吸收系数的影响
在TCDD分子基态几何结构优化的基础上,采用TDDFT方法研究了0—0.025 a.u.的外电场对分子前26个激发态的波长λ和摩尔吸收系数的影响,如图4所示.从图中可以看出,当外电场较弱时,吸收峰的波长变化不明显,但随着外电场的增强,吸收峰出现显著红移,当外电场增大到0.02 a.u.时,红移已经非常明显,这是由于当外电场较弱时,电子云左右对称分布在分子上(如图5所示),苯环左右两侧Cl原子的电子云密度相当;随着外电场的增强,分子中的电子发生了整体转移,苯环上的电子沿电场的方向偏移,苯环右侧Cl原子的电子云密度增加,苯环左侧Cl原子的电子云密度明显减小,如图5中的占据轨道所示;同理,电子的整体转移也使得分子跃迁轨道中苯环上的电子云密度减小,苯环左侧Cl原子上电子云密度剧增,如图5中的跃迁轨道所示.外电场引起的电子云偏移也使得苯环的大π键变弱,π→π∗跃迁的能量降低,跃迁产生的波长增大,即吸收峰红移.当外电场增大到0.02 a.u.时,分子的占据轨道和跃迁轨道的电子云偏移现象已很明显,因此,此时的吸收峰红移现象也已非常显著.
随着外电场的增强,吸收峰的红移,摩尔吸收系数开始降低,当外电场增强到0.02 a.u.时,摩尔吸收系数降低已经非常显著,这是由于外电场对电子的整体偏移,使得苯环及其周围的电子云密度减小(如图5所示),因此π→ π∗跃迁的电子个数减少,从而摩尔吸收系数降低.
5 结 论
1)本文采用DFT方法,对不同外电场下TCDD分子几何结构的变化进行了研究.结果表明,分子结构和分子总能量对外电场有着强的依赖关系.
2)采用TDDFT方法,对不同外电场下TCDD分子激发态波长和摩尔吸收系数的变化进行了研究.结果表明,无外电场时,分子最强吸收峰的波长为221 nm,由环状共轭的三个乙烯键的苯型体系中π→π∗电子跃迁所产生;随着外电场的增强,苯环上的电子云密度减小,吸收峰红移明显,摩尔吸收系数剧烈下降.
综上分析可见,在外电场的作用下,TCDD的分子结构变化剧烈.本文工作对TCDD的检测和降解方法研究提供了一定的理论依据,也对其他环境毒物的检测方法和降解机理研究有启示作用.
猜你喜欢
中学化学(2022年5期)2022-06-17
数学物理学报(2022年3期)2022-05-25
数学物理学报(2022年1期)2022-03-16
数学物理学报(2021年5期)2021-11-19
华南师范大学学报(自然科学版)(2021年4期)2021-08-30
数学物理学报(2021年3期)2021-07-19
汕头大学学报(自然科学版)(2020年4期)2020-12-14
江苏理工学院学报(2020年2期)2020-10-23
理科考试研究·高中(2019年8期)2019-09-19
化学教学(2015年11期)2015-12-19
