
硫辛酸酯类衍生物的合成方法学研究
2018-12-04 07:43张露云
通化师范学院学报 2018年12期
张露云,徐 倩,丛 丽,王 雨,臧 皓
硫辛酸(lipoic acid),亦称为α-硫辛酸,是一种天然的二硫化合物,广泛分布于动植物的组织中[1],是一种内源性抗氧化剂,最初于1950年由美国的Reed[2]等从猪肝中分离提取得到,硫辛酸可以穿过血脑屏障进入脑内[3].同时在几种生化过程中发挥重要作用,作为抗氧化剂,硫辛酸可以清除单线态氧[4]、过氧亚硝基[5]、过氧自由基、羟基自由基和次氯酸[6]等各种活性氧,在临床上主要用于治疗糖尿病周围神经病变[7]、阿尔茨海默症[8]和炎症[9].硫辛酸几乎没有毒副作用,不仅可以作为药品和保健品,还可以用于饲料添加剂及化妆品中,近年来受到广泛关注.
虽然硫辛酸口服吸收迅速,但肝首过效应明显,平均绝对生物利用度约为30%[10],代谢很快,平均血浆消除半衰期约为0.5h[11],服用量为每日600mg,造成浪费的同时还增加了身体的负担,因此对其进行结构修饰研究是药物化学领域的研究热点.Cravero等[12]采用1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐和1-羟基苯并三唑为缩合剂,甲醇和N,N-二甲基甲酰胺(1∶1)为溶剂,室温反应22h得到硫辛酸甲酯,该反应时间较长,N,N-二甲基甲酰胺沸点高难以除去.Koufaki等[13]采用氯化亚砜活化硫辛酸羧基后与乙醇反应得到硫辛酸乙酯,该方法所使用的氯化亚砜刺激性强,难于保存,而且反应产生氯化氢和二氧化硫气体腐蚀设备,污染环境.Lawrence等[14]采用乙酰氯和乙醇反应2h后再加入硫辛酸的策略合成硫辛酸乙酯,该反应同样产生氯化氢气体.因此,建立一种合成硫辛酸酯类衍生物的新方法具有重要意义.
1 实验部分
1.1 试剂与仪器
硫辛酸(纯度98%)、三苯基膦(TPP,纯度98%)、偶氮二甲酸二异丙酯(DIAD,纯度97%)、二环己基碳二亚胺(DCC,纯度98%)、4-二甲氨基吡啶(DMAP,纯度98%)、N-羟基丁二酰亚胺(NHS,纯度98%)、无水四氢呋喃(纯度99.5%),均购自萨恩化学技术有限公司;硫酸(H2SO4)购自天津科密欧化学试剂有限公司;其他试剂和溶剂均为市售分析纯.
QUINTIX35-1CN型电子分析天平(德国赛多利斯公司);Bruker-600型核磁共振波谱仪(TMS为内标,德国Bruker公司);Q-TOF-MS6520型质谱仪(美国Agilent公司);DFY-5型低温反应浴槽(巩义市英峪高科仪器厂);2XZ-2C型真空泵(临海市谭氏真空设备有限公司).
1.2 实验过程
(1)硫辛酸丙酯(3a)的合成路线.采用四条路线合成化合物3a,具体操作方法如下:
路线①:将硫辛酸(0.6mmol)、丙醇(0.6mmol)放入干燥的Schlenk管中,加入THF(1.5mL),再加入H2SO4(150μL),室温反应12h后薄层色谱检测.
路线②:将硫辛酸(0.6mmol)、丙醇(0.6mmol)、TPP(0.6mmol)于 N2条件下放入干燥的 Schlenk管中,加入 THF(1.5mL),于 0℃下加入 DIAD(0.6mmol),室温反应24h后薄层色谱检测.
路线③:将硫辛酸(0.6mmol)、DCC(0.6mmol)、NHS(0.6mmol)放入干燥的 Schlenk 管中,加入THF(1.5mL)后活化12h,随后加入丙醇(0.6mmol),室温反应12h后薄层色谱检测.
路线④:将硫辛酸(0.6mmol)、DCC(0.6mmol)、DMAP(0.06mmol)放入干燥的Schlenk管中,加入THF(1.5mL)后活化12h,随后加入丙醇(0.6mmol),室温反应12h后薄层色谱检测.
经过分析薄层板,确定路线①为化合物3a的合成路线,现将合成路线进行工艺优化.
(2)合成方法的确立.
首先是反应温度考察.在路线①的基础上对反应温度进行考察,分别考察了反应温度为20℃、40℃、50℃、60℃和70℃时3a的收率.由图1可知,随着反应温度的上升,3a收率呈先上升后平稳的趋势,因此选择比较温和的50℃作为反应温度.
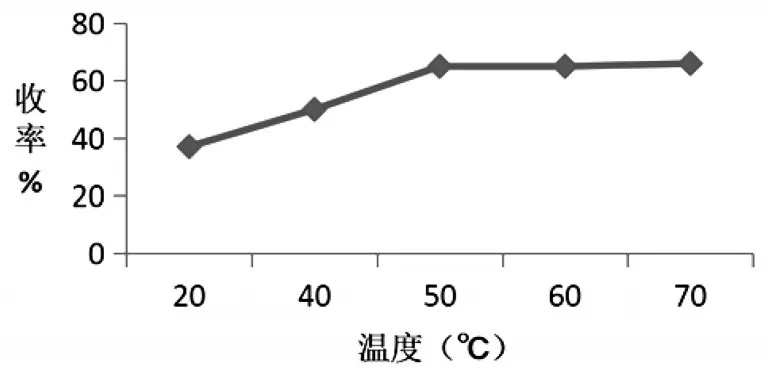
图1 不同反应温度对3a收率的影响
其次是投料比考察.在路线①的基础上,固定了反应温度为50℃,对丙醇和硫辛酸的投料比(mol∶mol)进行考察,分别考察了投料比为1∶1、1.3∶1、1.5∶1、2.0∶1和2.5∶1时3a的收率.由图2可知,随着投料比的增大,收率呈先上升后下降的趋势,最后确定反应投料比为2.0∶1.
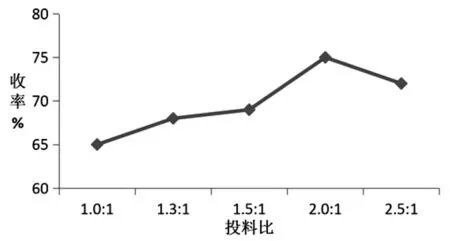
图2 不同反应投料比对3a收率的影响
然后是反应时间考察.在路线①的基础上,固定了反应温度为50℃,投料比为2.0∶1,对反应时间进行考察,分别考察了3h、6h、12h、18h和24h时3a的收率.由图3可知,收率呈先上升后平稳的趋势,因此选择时间最短的6h作为反应时间.
最后是催化剂用量考察.在路线①的基础上,固定反应温度为50℃,投料比为2.0∶1,反应时间为6h,对催化剂用量进行考察,分别考察了硫酸用量为25μL、50μL、100μL、150μL和200μL时3a的收率.由图4可知,收率呈先上升后下降的趋势,因此选择收率最高时的催化剂用量50μL.
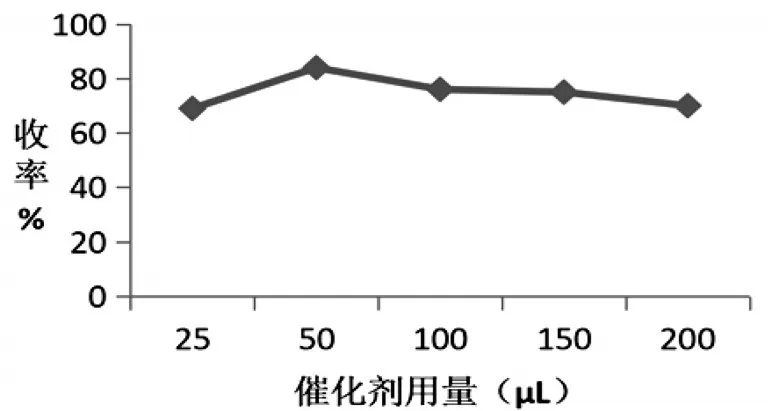
图4 不同催化剂用量对3a收率的影响
(3)合成方法的应用.应用建立的合成方法合成了硫辛酸丙酯(3a)、硫辛酸丁酯(3b)、硫辛酸己酯(3c)、硫辛酸庚酯(3d)和硫辛酸壬酯(3e),合成路线见图5,收率分别为84%、89%、80%、91%、69%,化合物3a~3e经核磁共振波谱和高分辨质谱确证均为目标化合物,数据如下.
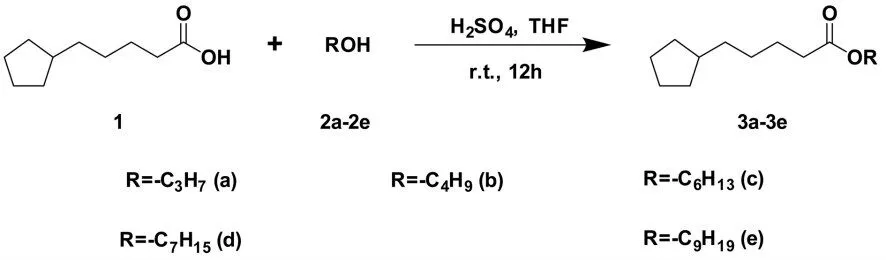
图5 化合物3a~3e的合成路线
化合物3a,黄色油状物,收率84%.1H-NMR(DMSO-d6,600MHz,ppm):δ 3.96(s,2H),3.61(q,1H),3.18(q,1H),3.12(q,1H),2.41(d,1H),2.30(s,2H),1.86(q,1H),1.67(d,1H),1.57(s,5H),1.38(d,2H),0.88(s,3H).13C-NMR(DMSO-d6,150MHz,ppm):δ 172.8,65.1,56.0,39.9,38.1,34.0,33.3,28.1,24.2,21.5,10.2;MS m/z:271.0683(M+Na).
化合物3b,黄色油状物,收率89%.1H-NMR(DMSO-d6,600MHz,ppm):δ 4.01(s,2H),3.60(q,1H),3.18(d,1H),3.12(s,1H),2.41(d,1H),2.29(s,2H),1.87(t,1H),1.66(s,1H),1.54(s,5H),1.33(t,4H),0.88(s,3H).13C-NMR(DMSO-d6,150MHz,ppm):δ 172.8,63.6,56.0,39.9,38.1,34.0,33.3,30.2,28.1,24.2,18.6,13.5;285.1959(M+Na).
化合物3c,黄色油状物,收率80%.1H-NMR(DMSO-d6,600MHz,ppm):δ 4.00(s,2H),3.60(q,1H),3.18(t,1H),3.11(t,1H),2.41(d,1H),2.29(s,2H),1.86(q,1H),1.66(s,1H),1.55(s,5H),1.37(d,2H),1.27(s,6H),0.86(s,3H).13CNMR(DMSO-d6,150MHz,ppm):δ 172.8,63.7,56.0,39.9,38.1,34.0,33.3,30.8,28.1,28.1,25.0,24.2,22.0,13.8;MS m/z:313.2271(M+Na).
化合物3d,黄色油状物,收率91%.1H-NMR(DMSO-d6,600MHz,ppm):δ 4.00(s,2H),3.60(t,1H),3.17(s,1H),3.12(s,1H),2.41(d,1H),2.29(s,2H),1.86(q,1H),1.66(t,1H),1.54(d,5H),1.37(d,2H),1.26(d,8H),0.86(s,3H).13CNMR(DMSO-d6,150MHz,ppm):δ 172.8,63.6,56.0,39.9,38.1,34.0,33.3,31.1,28.3,28.1,28.0,25.3,24.2,22.0,13.9;MS m/z:327.2429(M+Na).
化合物3e,黄色油状物,收率69%.1H-NMR(DMSO-d6,600MHz,ppm):δ 4.00(s,2H),3.60(q,1H),3.17(d,1H),3.12(s,1H),2.41(d,1H),2.29(s,2H),1.87(d,1H),1.66(q,1H),1.54(d,5H),1.37(d,2H),1.26(t,12H),0.86(s,3H).13CNMR(DMSO-d6,150MHz,ppm):δ 172.7,63.6,56.0,39.8,38.0,34.0,33.3,31.2,28.8,28.6,28.6,28.1,28.0,25.3,24.2,22.0,13.9;MS m/z:355.2741(M+Na).
2 讨论
2.1 化合物3a合成路线的确定
上述四种合成路线反应结束后采用薄层色谱法检测,通过对原料与产物的比对分析,确认路线①、路线③和路线④共有点为目标点,将路线①反应液蒸干,硅胶柱层析分离得到黄色油状物,经核磁共振氢谱(见图6)和高分辨质谱(见图7)确证为目标化合物.
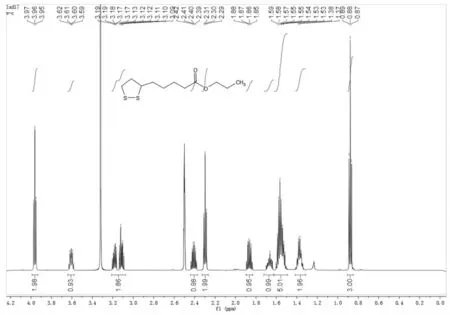
图6 化合物3a的核磁共振氢谱图
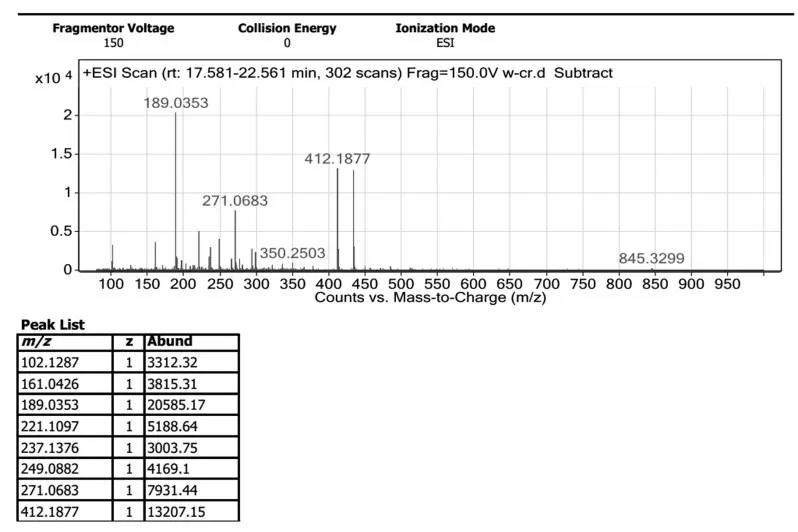
图7 化合物3a的高分辨质谱图
从薄层板上分析,路线②未生成3a,因此首先放弃;路线③生成了3a,但是从薄层板上判断其收率不高,NHS一般制备酰胺时比较常用,分析原因可能是在制备酯类化合物时由于醇羟基活性较弱,无法与生成的活性酯反应导致目标化合物产生较少,故放弃此方法;路线④也生成了目标化合物,但是生成的杂质点与目标化合物难以分开,因此未选用此路线;路线①在薄层板上只有两个斑点,杂质较少,斑点间距较远,易于纯化,且收率较高,因此综合考虑,选用路线①作为3a的合成路线.
2.2 化合物3a合成路线的优化
通过单因素实验,分别对其反应温度、投料比、反应时间、催化剂用量进行考察.反应温度考察结果显示,随着反应温度的上升,收率呈先上升后平稳的趋势,所以在反应时,应把反应温度控制在50℃,避免温度过高产生杂质而造成收率下降.投料比考察结果显示,随着投料比的增大,收率呈先上升后下降的趋势,从收率和节省原料的角度考虑,确定投料比为2.0∶1.反应时间考察结果显示,收率呈先上升后平稳的趋势,因此选择反应时间为6h.催化剂用量考察结果显示,收率呈先上升后下降的趋势,考虑是催化剂用量过大副反应增加所致,考虑到催化剂的消耗,本文以50μL作为适宜的催化剂用量.
综上,化合物3a的最佳反应条件为反应温度为50℃、投料比为2.0∶1、反应时间为6h、催化剂用量为50μL.
3 结论
本研究建立的合成方法具有反应时间短、收率高、反应条件较温和等优点,为硫辛酸酯类衍生物的合成提供了新的方法和依据.
猜你喜欢
粮食加工(2022年1期)2022-03-23
玻璃(2022年1期)2022-02-23
水泵技术(2021年4期)2021-01-22
中华养生保健(2020年4期)2020-11-16
铜仁学院学报(2018年6期)2018-07-05
计算机系统应用(2017年5期)2017-06-07
海峡科技与产业(2016年3期)2016-05-17
中华老年多器官疾病杂志(2016年7期)2016-04-28
中国当代医药(2015年8期)2015-03-01
中国卫生标准管理(2015年7期)2015-01-27
