
CH2CH2OH与HO2反应机理及动力学性质的理论研究
2018-12-03 00:19文明杰姚秋月
西北师范大学学报(自然科学版) 2018年6期
许 琼,文明杰,姚秋月,李 娜,王 睿
(陕西理工大学化学与环境科学学院,陕西省催化基础与应用重点实验室,陕西汉中 723000)
乙醇作为当前主要的可再生能源,吸引着越来越多的研究人员的关注.乙醇裂解反应和氧化反应的主要中间体是CH3CH2O、CH3CHOH及CH2CH2OH自由基[1-2],反应如下:
这些自由基在碳氢化合物的链增长反应中扮演着不可或缺的角色[3].此外,CH2CH2OH作为一种活性自由基,可以发生异构转化为CH3CHOH和CH3CH2O,同时也可以和多种小分子物种发生大气化学反应和燃烧反应[4-5].因此,研究CH2CH2OH自由基氧化过程的机理和动力学,对深刻了解大气中含氧自由基的形成、污染防治,以及星际物种变迁有着重要的理论和实际意义[6-9].
HO2是表征大气氧化能力的关键物种,同时也是碳氢燃料燃烧反应、大气光化学反应及一些生化反应中非常重要的高活性瞬时中间体.实验研究表明,CH2CH2OH能被HO2快速氧化生成稳定物种O2H2O和H2等[10-12].因此,CH2CH2OH与HO2反应的机理研究对大气自由基物种循环过程意义重大.
HO2参与的反应具有十分复杂的势能面信息.Anglada等[13]对HO2+HO2/CH3O2系列反应进行了理论研究,并指出这些反应都以三重态抽氢反应为主.Mousavipour等[14]指出CH3O+HO2反应也以三重态抽氢反应为主.然而,CH3O的异构体CH2OH和HO2[15]的反应则以单重态碳氧耦合机理为主.对于CH2CH2OH与HO2反应的机理研究至今还未见报道,为了进一步完善HO2反应的势能面信息,文中选取图1所示的CH2CH2OH与HO2反应体系研究其单、三重态反应机理,并在此基础上计算了各通道的反应速率常数,为揭示HO2反应规律提供有意义的理论依据和参考.
1 计算方法
在B3LYP/CBSB7水平下全参数优化了反应物、过渡态、中间体及产物等各驻点物种的几何构型.通过对振动频率的分析,确认了各驻点物种的几何构型,并在相同水平下进行内禀反应坐标(IRC)分析,验证了各反应路径中过渡态与相应反应物(前中间体)和产物(后中间体)的相关性[16-18].为了获得更可靠的能量信息,采用CBS-QB3方法对B3LYP/CBSB7水平上优化的所有驻点物种进行了单点能校正.文中所涉及的电子结构计算均使用Gaussian09程序完成.
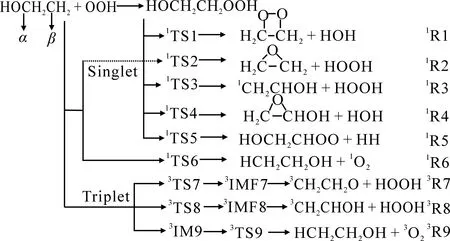
图1 CH2CH2OH+HO2在单、三重态的反应通道
在获得反应体系各驻点物种的能量相关信息后,使用VKLab程序包,采用传统过渡态理论(TST)和稳态近似法计算CH2CH2OH+HO2反应的动力学性质.
如(4)式所示,主要标题反应从一个前中间体开始,形成过渡态,最终生成产物:
近似反应物的平衡状态下,整体速率常数表示为
(5)
如果kuni≪k-1,速率常数可表示为
(6)
其中反应第二步速率常数kuni通过VKLab程序给出,Keq由下式计算得出:
(7)
其中,不同的Q值分别为中间体、反应物R1和反应物R2的配分函数,所有的配分函数都在B3LYP/CBSB7水平上计算;ER,ECR分别为反应物和中间体的总能量;σ为对称因子.
2 结果与讨论
计算结果显示,在CBS-QB3水平上CH2CH2OH中(β-C)原子的自旋密度为1.05[19].因此可以推测,CH2CH2OH上未配对的电子主要集中在(β-C)上.此外,在相同水平下,测得HO2中端基O原子的自旋密度[20-22]是0.74,表明在HO2上未配对的电子主要集中在端基O上.在CH2CH2OH+HO2中,主要发生的方式如下:
(a)HO2中的端基(非端基)O抽取CH2CH2OH羟基中的H原子;
(b)HO2中的端基(非端基)O抽取CH2CH2OH(α-C)上的H原子;
(c)CH2CH2OH中(β-C)抽取HO2中的H.
基于上述分析,推测CH2CH2OH+HO2反应中存在9条可能的反应通道(图1),其中单重态有6条(R1~R6),三重态存在3条(R7~R9).
表1给出了CBS-QB3水平上CH2CH2OH+HO2反应中各驻点物种的能量信息,相对能ΔE/(kJ·mol-1)、焓ΔH(298)/(kJ·mol-1)、吉布斯自由能ΔG(298)/(kJ·mol-1)、熵S/(kJ·mol-1·K-1)、过渡态虚频ν/cm-1以及自旋角动量平方算符本征值S2[23-25].如表1所示,CBS-QB3水平上双重态物种CH2CH2OH和HO2其自旋角动量平方算符本征值S2为0.750,三重态物种3CH3CHO,3CH2CH2O,3CH2CHOH和3O2其自旋角动量平方算符本征值S2为2.000,而双重态和三重态消除自旋污染[26]后的本征值分别为0.750与2.000,这与优化后的本征值完全吻合,说明用CBS-QB3方法研究标题反应时波函数基本没有自旋污染.由此证实了文中所选用的CBS-QB3方法是合理的.
2.1 单重态反应机理
如图2所示,CH2CH2OH+HO2反应在单重态上的通道(R1,R3,R4和R5)分别开始于同一个中间体,中间体的稳定化能为-262.67 kJ·mol-1.
表1 CH2CH2OH+HO2反应体系中所有物种的相对能ΔE、焓ΔH、吉布斯自由能ΔG(298)、熵S、过渡态虚频ν以及自旋角动量平方算符本征值S2
Tab 1 Energies ΔE,enthalpies ΔH(298),free energies ΔG(298),entropies S,virtual frequencies ν and the S2 after spin of all the species for CH2CH2OH+HO2 reaction
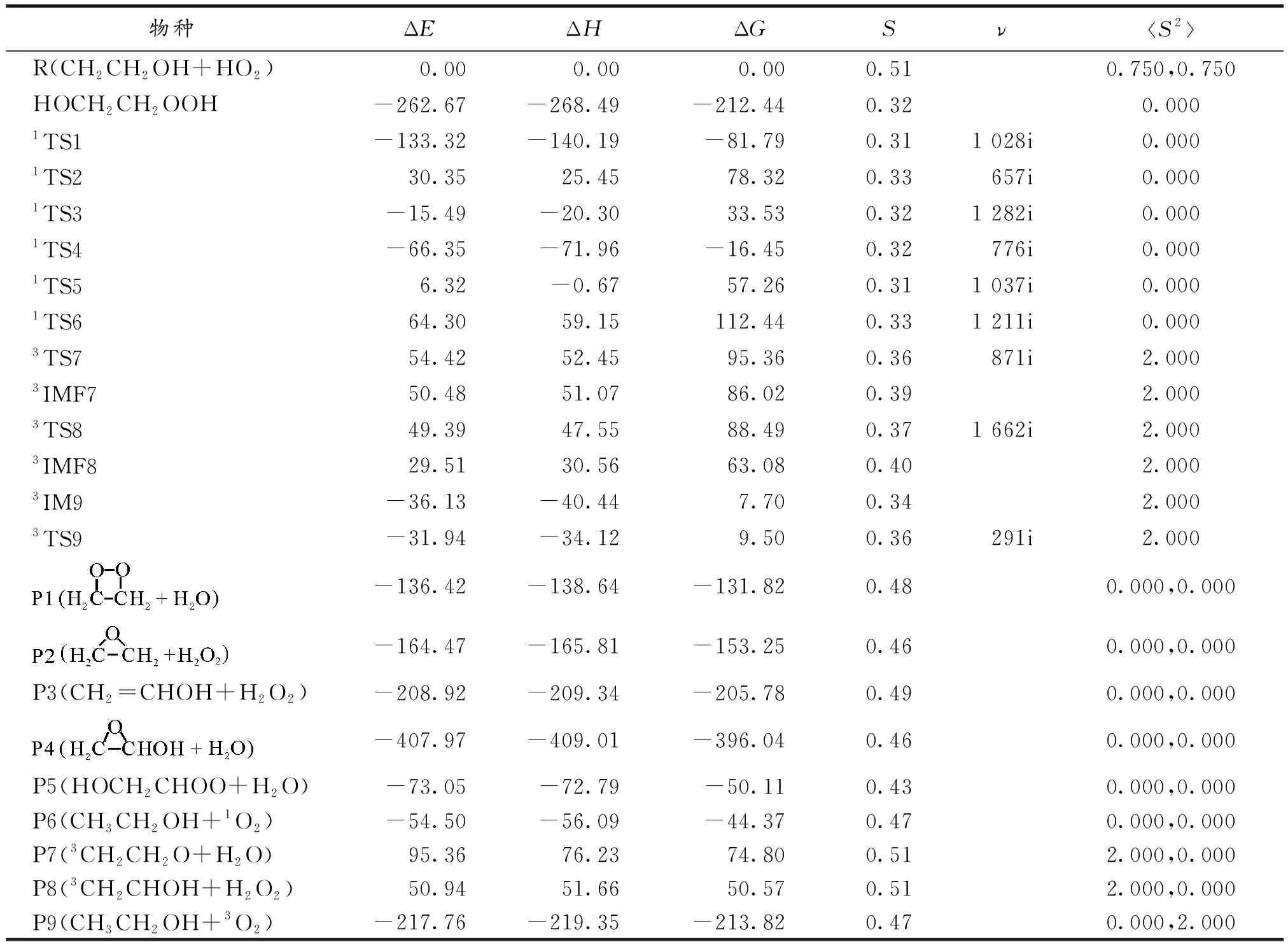
表1 CH2CH2OH+HO2反应体系中所有物种的相对能ΔE、焓ΔH、吉布斯自由能ΔG(298)、熵S、过渡态虚频ν以及自旋角动量平方算符本征值S2
物种ΔEΔHΔGSνS2R(CH2CH2OH+HO2) 0.00 0.00 0.000.510.750,0.750HOCH2CH2OOH-262.67-268.49-212.440.320.0001TS1-133.32-140.19-81.790.311 028i0.0001TS230.3525.4578.320.33657i0.0001TS3-15.49-20.3033.530.321 282i0.0001TS4-66.35-71.96-16.450.32776i0.0001TS56.32-0.6757.260.311 037i0.0001TS664.3059.15112.440.331 211i0.0003TS754.4252.4595.360.36871i2.0003IMF750.4851.0786.020.392.0003TS849.3947.5588.490.371 662i2.0003IMF829.5130.5663.080.402.0003IM9-36.13-40.447.700.342.0003TS9-31.94-34.129.500.36291i2.000-136.42-138.64-131.820.480.000,0.000-164.47-165.81-153.250.460.000,0.000P3(CH2=CHOH+H2O2)-208.92-209.34-205.780.490.000,0.000-407.97-409.01-396.040.460.000,0.000P5(HOCH2CHOO+H2O)-73.05-72.79-50.110.430.000,0.000P6(CH3CH2OH+1O2)-54.50-56.09-44.370.470.000,0.000P7(3CH2CH2O+H2O)95.3676.2374.800.512.000,0.000P8(3CH2CHOH+H2O2)50.9451.6650.570.512.000,0.000P9(CH3CH2OH+3O2)-217.76-219.35-213.820.470.000,2.000
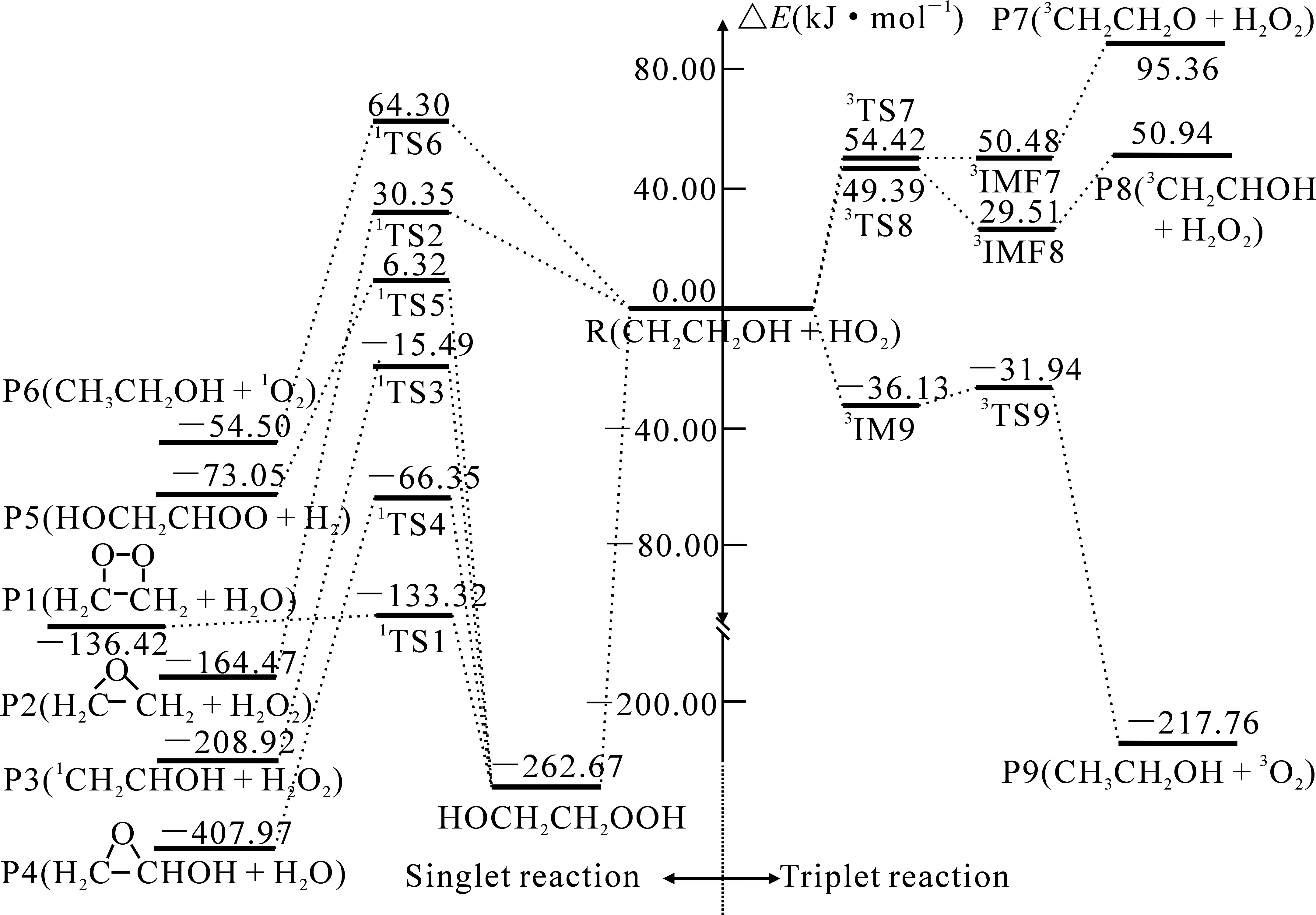
图2 CBS-QB3水平上计算得到的CH2CH2OH+HO2反应单、三重态的反应势能剖面(能量:kJ·mol-1)
通道R6(R→1IM6→1TS6→P6)经过1TS6,H(1)在C(1)和O(1)之间振动,紧接着H(1)—O(1)键断裂,CH2CH2OH中(β-C)上有一个不成对电子,使得H(1)原子转移到(β-C)原子上,协同反应生成P6(CH3CH2OH+1O2).通道R6经历的1TS6具有较高的表观活化能,与通道R1相比,通道R6更难进行.
2.2 三重态反应机理
反应CH2CH2OH+HO2在三重态有3种反应通道,分别为通道R7,R8和R9.
对于通道R9(R→3IM9→3TS9→P9),从图3各物种的几何构型可以看出,CH2CH2OH中(β-C)原子与HO2上的H原子通过氢键作用形成一个中间体3IM9,其中氢键键长为0.186 nm,随后H(1)—C(1)原子间作用逐渐增强,而H(1)—O(1)原子间作用减弱,经过3TS9,H(1)在C(1)和O(1)之间进行振动,最终H(1)与O(1)键断裂,CH2CH2OH中(β-C)原子抽取HO2中的H原子生成产物P9(CH3CH2OH+3O2).对于通道R9(R→3IM9→3TS9→P9)需克服能垒4.19 kJ·mol-1.与单重态生成产物P6(CH3CH2OH+1O2)的通道R6相比,通道R9需跨过的反应能垒较低,且经过的过渡态表观活化能较低,通道R6相对于通道R9来说更难进行.
对于通道R7(R→3IM7→3TS7→P7),通过R7经过3TS7,H(2)—O(2)键作用逐渐增强,H(2)—O(3)键作用减弱,其中CH2CH2OH中羟基部分的H原子与HO2上的端基O原子通过氢键作用形成一个后中间体3IMF7,H(2)在O(2)与O(3)之间进行振动,最终导致H(2)—O(3)键断裂,HO2上的端基O原子抽取CH2CH2OH中羟基上的H原子生成产物P7(3CH2CH2O+H2O).与单重态生成产物P2的通道R2相比,通道R7的过渡态表观活化能较高,且通道R2为放热反应,所以通道R7比通道R2更难进行.通道R7需经过的过渡态表观活化能较高且为吸热反应,为动力学禁阻反应.
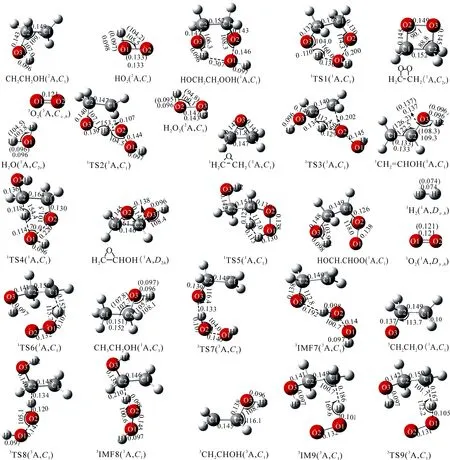
图3 表示CH2CH2OH+HO2反应中反应物、产物、中间体和过渡态在CBS-QB3理论水平上优化后得到的几何构型和可获得的实验数据
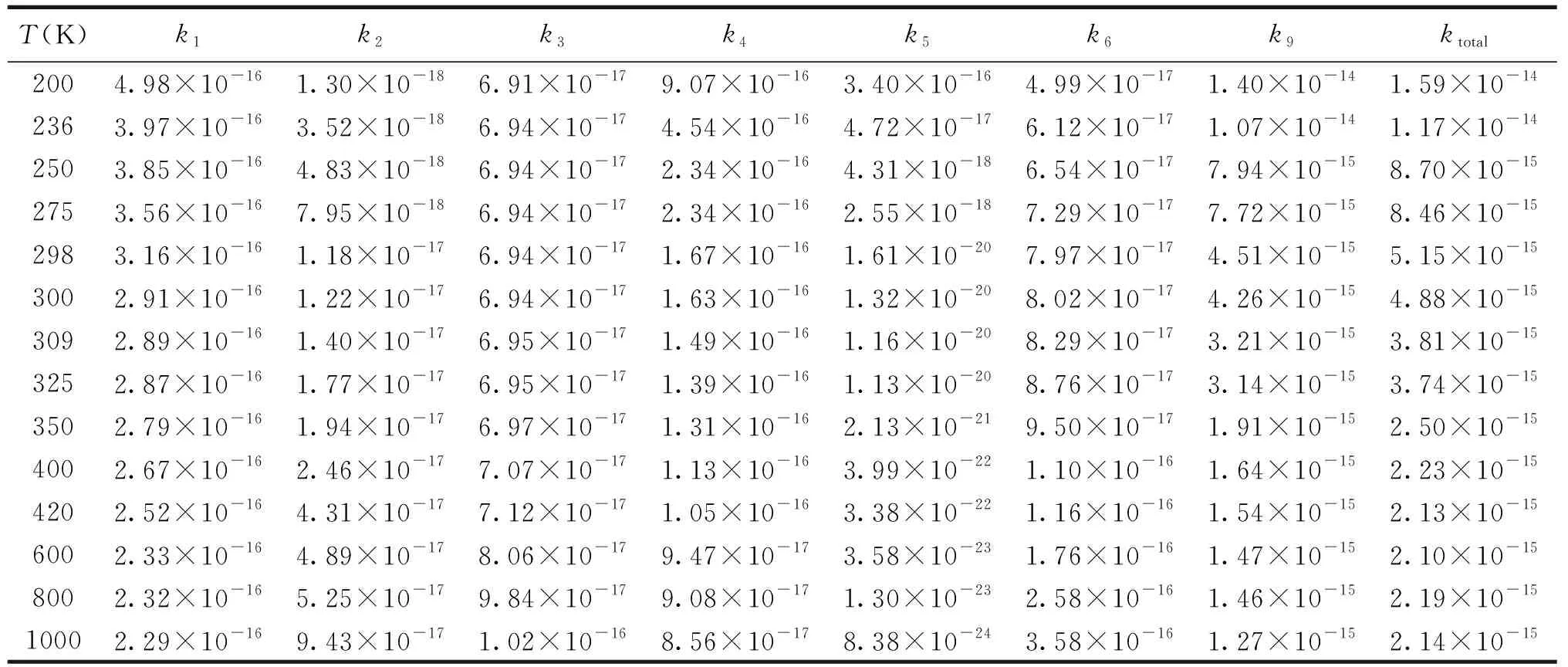
表2 通道R1,R2,R3,R4,R5,R6和R9在200~1 000 K温度范围内速率常数k/(cm3·molecule-1·s-1)
对于通道R8(R→3IM8→3TS8→P8),经过3TS8,H(3)—O(2)键作用逐渐增强,H(3)—C(2)键作用减弱,其中CH2CH2OH中α-C原子上的H与HO2上的端基O原子通过氢键作用形成一个后中间体3IMF8,H(3)在O(2)与C(2)之间进行振动,最终导致H(3)—C(2)键断裂,HO2上的端基O原子抽取CH2CH2OH中α-C原子上的H原子生成产物P8(3CH2CHOH+H2O2).与单重态生成产物P3(1CH2CHOH+H2O2)的通道R3相比,通道R8经过的过渡态表观活化能较高,且通道R3为放热反应,所以通道R8比通道R3更难进行.通道R8需要经过的过渡态表观活化能较高且为吸热反应,为动力学禁阻反应.
分析得出,CH2CH2OH+HO2反应在三重态势能面上主要通过R9(R→3IM9→3TS9→P9(CH3CH2OH+3O2))进行.
2.3 主通道速率常数计算
在CBS-QB3水平上对CH2CH2OH+ HO2反应机理和速率常数进行研究.表2给出了标题反应单重态反应通道R1~R6、三重态反应通道R9在200~1 000 K内各路径速率常数k.图4为200~1 000 K内速率常数k随温度的变化曲线图.
由表2及图4知,速率常数k1,k4,k5和k9与温度具有负温度系数效应;k2,k3,k6则与温度具有正温度系数效应,其中k5随温度的升高,变化幅度较明显.标题反应优势通道R9的速率常数k在200~1 000 K内具有负温度系数效应.
为了方便讨论,在得出各个通道速率常数的基础上,计算了如图5 所示的随温度升高各个通道速率常数分支比的数值.由图5可知,通道R9(R→3IM9→3TS9→P9)的速率在整个温度范围内占有绝对优势,一直保持在59%以上.通道R9(R→3IM9→3TS9→P9)与通道R1(R→1IM1→1TS1→P1)在高温区为竞争反应.k2,k3,k4和k5在整个温度变化过程中变化幅度不大,所占比例集中在0~10%之间.
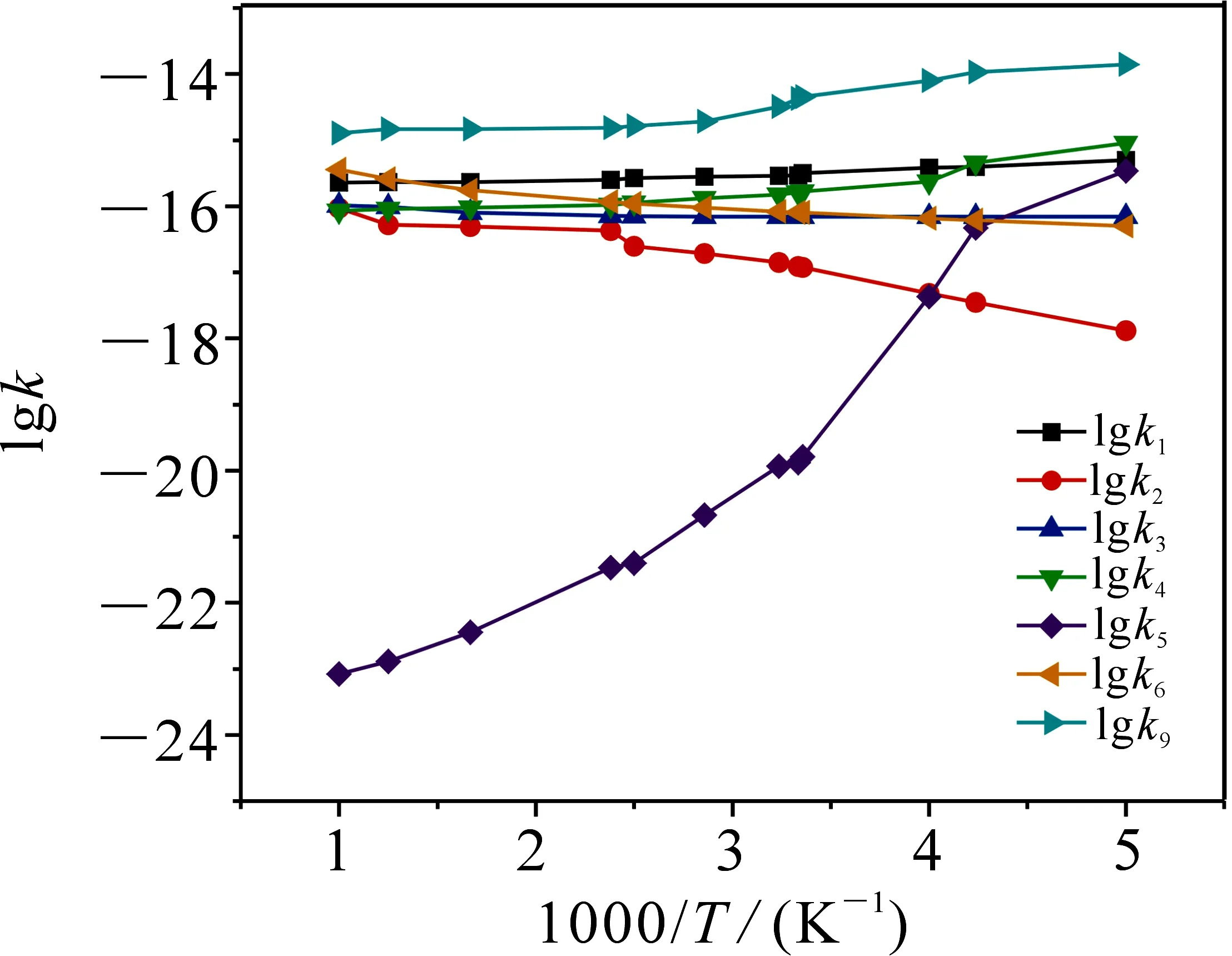
图4 200~1 000 K温度范围内速率常数k随温度的变化曲线图
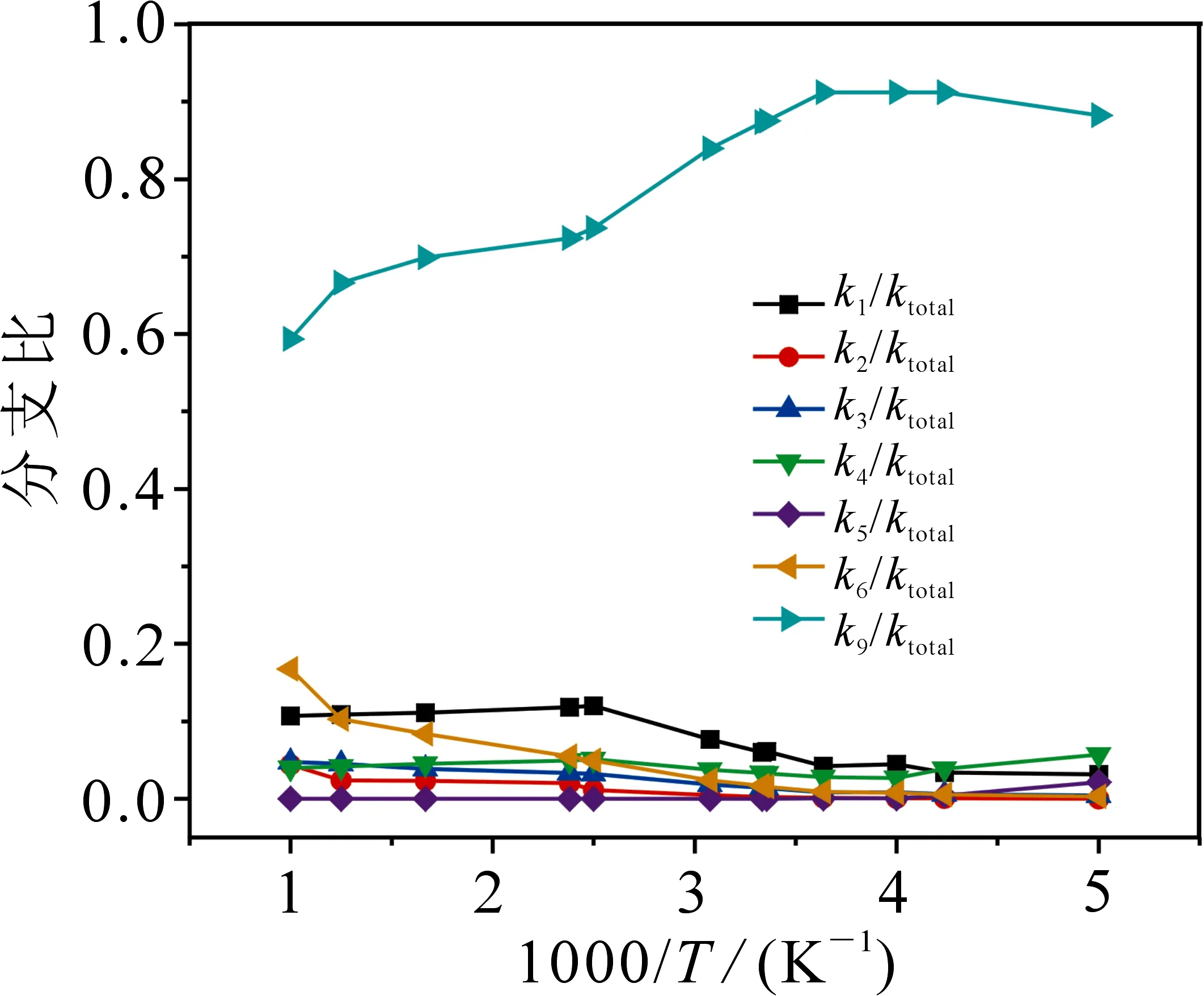
图5 200~1000 K范围内分支比随温度的变化
3 结论
在CBS-QB3水平上对CH2CH2OH+HO2反应机理及动力学性质进行了详细探讨,给出了反应势能面和主通道速率常数,得到了以下结论:
2)比较单、三重态势能面信息可以得出,反应CH2CH2OH+HO2主要发生在三重态势能面上,生成产物P9(CH3CH2OH+3O2)的通道R9是整个反应的优势通道.
3)标题反应的优势通道R9(R→3IM9→3TS9→P9)的速率常数k在200~1000 K温度范围内具有负的温度系数效应.
猜你喜欢
北京航空航天大学学报(2022年5期)2022-06-06
中国药学药品知识仓库(2022年10期)2022-05-29
数学年刊A辑(中文版)(2021年1期)2021-06-09
汕头大学学报(自然科学版)(2020年4期)2020-12-14
电脑知识与技术(2018年3期)2018-03-21
中成药(2017年5期)2017-06-13
哈尔滨理工大学学报(2017年1期)2017-04-08
科技视界(2016年24期)2016-10-11
新高考·高一物理(2016年3期)2016-05-18
股市动态分析(2015年12期)2015-09-10
