
基于BC1群体的多性状QTL定位方法
2018-11-23 05:54:40胡家晶张小艺
绥化学院学报 2018年11期
佟 良 胡家晶 张小艺 孙 威
(绥化学院信息工程学院 黑龙江绥化 152061)
过去有很多科学工作者利用遗传标记的基本原理研究QTL,其中Jansen 1993给出了多个数量性状的区间作图方法[1]。Kaoet al.1999在CIM的基础上,发展形成一种新的作图方法——多区间定位方法MIM[2],该方法可同时对多个区间进行多个QTL作图,已被证明是比CIM更准确的估计方法。
之后,许多科学工作者在前人的基础上进一步推广,提出了一些适合特定情况的QTL分析方法,在其相应的范围内也有其作用。如考虑基因多效性和 (Q×E)交互,Jiang和Zeng 1995基于极大似然思想提出了多性状复合区间定位方法MT-CIM[3]。方法可同时对多个性状进行复合区间作图,相对于MIM能更好的提高QTL定位参数估计的精度。还有.Knott S A et al应用回归分析思想对多性状基因位点进行定位分析[4,5]。Xu et al 2005基于表型性状为多个离散的二进制对QTL进行了联合定位[6]。Guoet al 2007基于表型性状为不完整的数据对多性状QTL进行了定位研究[7]。Liu 2007与Banerjee et al.2008基于贝叶斯方法对多性状基因进行了研究[8,9]。Zeng 2012基于MIM思想,应用MT-CIM的模型提出了多性状多区间定位方法MT-MIM[10]。通过分析,MT-MIM方法的准确性高于MIM方法和MT-CIM方法。
多性状QTL定位方法,相对于单一性状QTL定位方法提高了检测功效、定位的精确度和估计功效。这是因为在多个表型性状条件下,单一性状QTL定位方法完全忽略了一个QTL对多个表型性状的共同影响。多性状QTL定位是关于多性状的一个QTL多效性检验,如果性状间存在较强的遗传相关,通过多性状QTL定位探测的功效是最高的[11].这些优势在动物[12,13]和植物[14]研究中得到广泛应用。
本文是在MT-MIM方法的基础上进一步拓展,给出了每个QTL基因型的重组率显性表达式,通过模拟对比MT-MIM方法,新方法对加性效应,显性效应的估计比MT-MIM方法要好。新方法通过重组率定位QTL在染色体上的位置明显好于MT-MIM方法。
一、背景知识与方法
(一)记号与背景。假定个杂交个体,个标记位点紧密连锁构成个标记区间,为表型性状个数。令,Yjt(j=1,…,n,i=1,…,t)表示第j个个体第i个表型性状值,X*jt(j=1,…,n,i=1,…,q+1)表示第j个个体第i个标记区间内的QTL基因型,Xji(j=1…,n,i=1,…,q+1)表示第j个个体第i个标记的标记基因型。我们对QTL基因型数字化
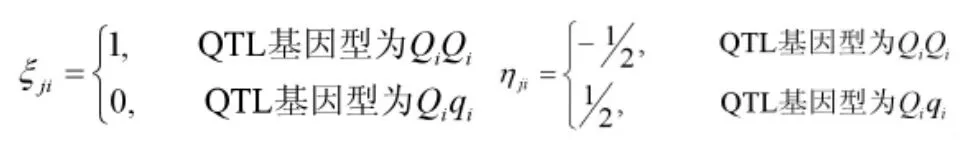
令ri和ri1分别表示第i个标记区间两侧标记之间的重组率和第i个标记区间内QTL与上标记位点之间的重组率。假设BC1群体每一个标记区间至多有一个QTL,第j个个体第个i标记区间已知时,其标记区间内QTL条件概率见表1
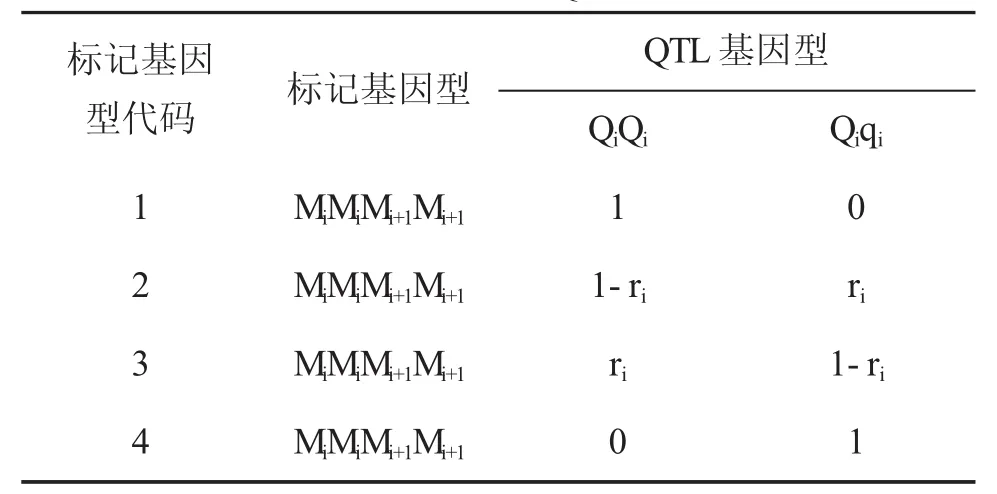
表1 标记基因型已知情况下QTL基因型的条件概率
(二)模型与似然函数。本文应用的统计模型是基于MIM思想,在MT-CIM模型的基础上提出的统计模型[15]
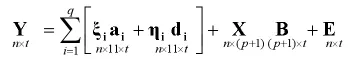
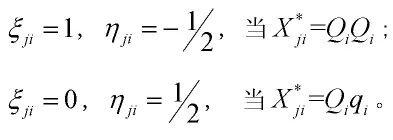
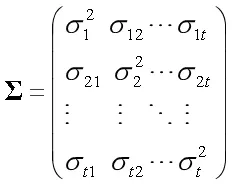
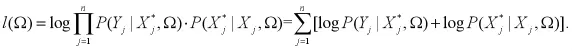
(三)EM算法。对于E步,考虑X,Y,Ω(S)我们计算l(Ω)的条件期望
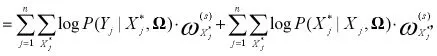
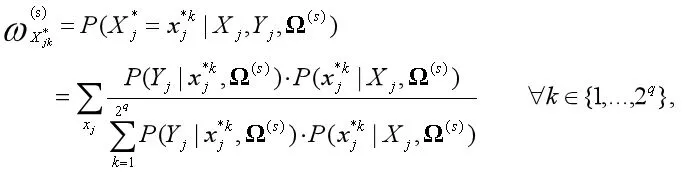
(四)参数估计。
1.C和Σ的估计。通过推导得出如下加性效应和显性效应的迭代表达式[15]
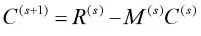
这里的 R(S)和 M(S)的表达式为

上式是R(S)对Joehanes[15]文章的修定,(Y-XB(S))'W(S)Di修定为D'iW'(S)(Y-XB(S)),i=1,...,cq。W=[ωx*jt]是一个n×2q的矩阵分别为第 i个 QTL第 k个表型性状加性效应和显性效应,D=(D1,D2,D3,…,Dcq),这里c的取值为1或者2。当只考虑加性效应时c=1,既考虑加性效应又考虑显性效应时c=2。当c=2时
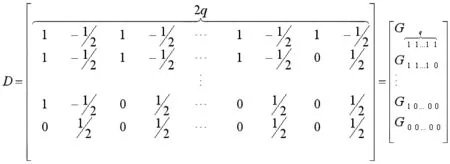
2.γ的估计。假定标记区间及其区间内的QTL具有关系表达式[16],
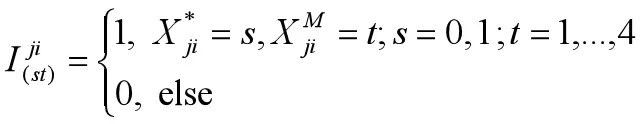
这里的j=1,...,n,i=1,...q.通过上面的示性函数可把Q函数第二部分和变形为如下形式
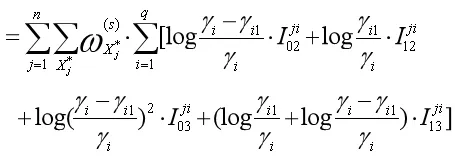
对上式进行极大化,可得重组率的显性表达式
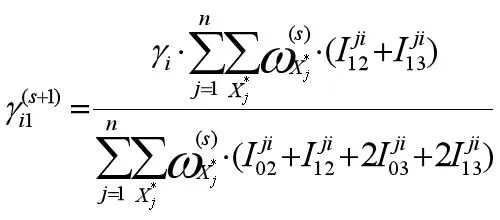
二、模拟与结果
为了评估给定参数估计方法的好坏,将本文提出的方法MT-MIM-BC1与MT-MIM进行对比。在不失一般性的情况下,模型中只考虑两个数量性状,2个QTL位点。由于估计的准确性随着标记区间的减小,逐渐提高,标记区间长度取定10cM。MT-MIM-BC1方法需要把区间长度10cM转换成重组率0.0906[17]。模型中参数选取使得遗传力在0.05和0.2附近。群体对QTL的影响很大,取定样本容量为500。为了评价估计的准确度,给出了每个参数的均方误差MSE。表2给出的是样本量为500,遗传力为0.2、0.05时MT-MIM-BC1方法与MT-MIM方法对QTL加性效应和显性效应的估计值和MSE。模拟结果显示在相同的遗传力条件下MT-MIM-BC1方法比MT-MIM方法的参数估计值更接近参数真值,MT-MIM-BC1方法对所有QTL加性效应和显性效应估计值的MSE和小于MT-MIM方法所对应的MSE和。说明MT-MIM-BC1方法比MT-MIM方法估计的精确度更高。随着遗传力的降低,两种方法的精准度都有所降低,但MT-MIM-BC1方法明显好于MT-MIM方法。

表2 h2=0.2,0.05,QTL效应的估计值
表3给出的是遗传力为0.05和0.2时QTL重组率估计值和两个表型性状协方差矩阵估计值以及相应参数MSE。QTL重组率是MT-MIM-BC1方法相对MT-MIM方法独有的。MT-MIM-BC1方法给出了QTL重组率的显性表达式,通过重组率能够更精准定位QTL在染色体上位置。MT-MIM 方法无法直接计算QTL重组率,表3中MT-MIM 方法重组率估计值是根据Joehanes[15]文章提供的方法重新编写的。程序中通过似然比LR定位QTL在染色体上位置,然后根据QTL位置及其所在区间上标记位点之间的距离得到重组率的估计值。从表3可以看出在相同的遗传力条件下MT-MIM-BC1方法重组率的估计值比MT-MIM 方法转化的重组率值更接近参数真值,估计的精度也比MT-MIM方法高。随着遗传力降低两种方法对参数估计的精准度逐渐变差,但MT-MIM-BC1方法对参数估计的精准度明显好于MT-MIM方法。两种方法对,,参数的估计值相差不大,但MT-MIM-BC1方法的MSE小于MT-MIM方法,说明MT-MIM-BC1方法参数估计精确度更高。
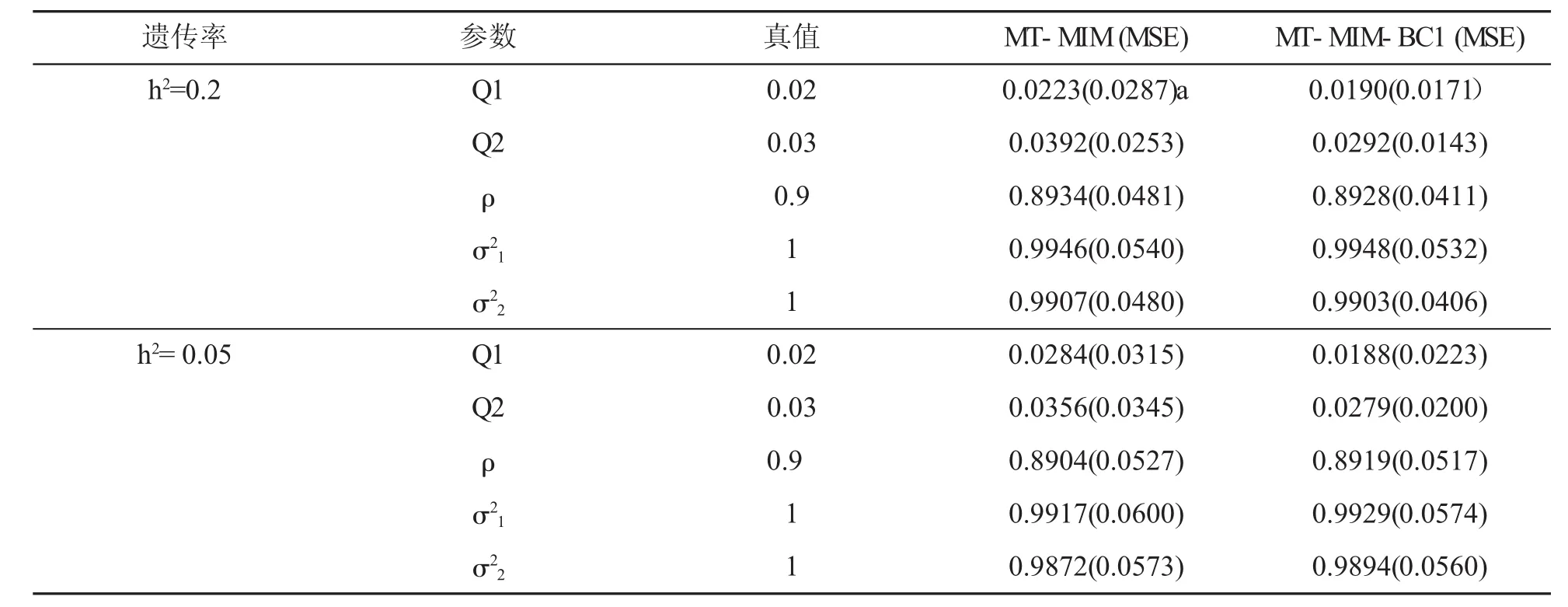
表3 h2=0.2,0.05,重组率和协方差估计值
MT-MIM-BC1方法选择的模型把非遗传标记位点效应也作为参数给出了其显性表达式。表4给出的是在遗传力为0.2和0.05条件下表型特征为T1和T2时非遗传标记位点效应真值和估计值以及MSE。模拟结果显示MT-MIM-BC1方法参数估计值比MT-MIM方法参数估计值更接近参数真值,MT-MIM-BC1方法所有标记位点效应的MSE和小于MT-MIM方法相应的MSE和。说明MT-MIM-BC1方法对标记位点效应估计的精确度高于MT-MIM方法。
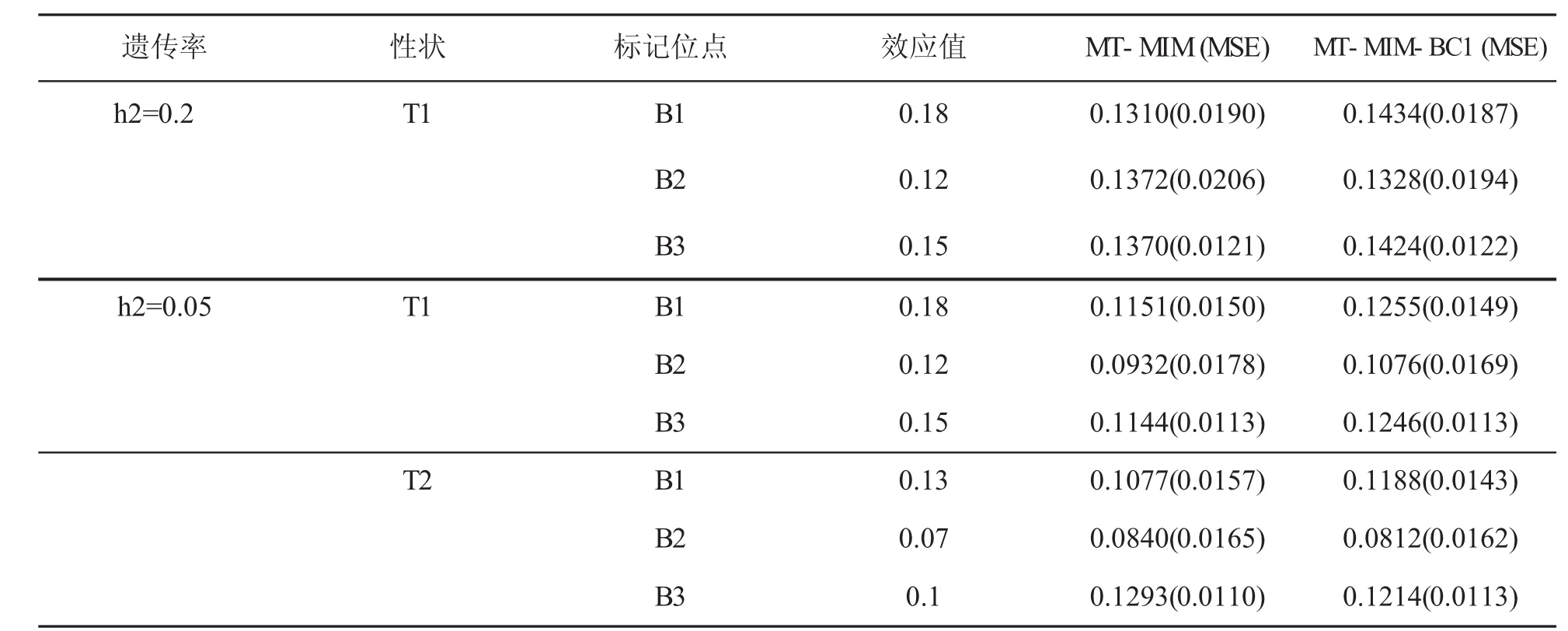
表4 h2=0.2,0.05,标记位点效应和MSE
三、结论与讨论
本文是在MT-MIM方法的基础上对BC1群体进行多性状多区间定位(MT-MIM-BC1)。在不失一般性的条件下,我们对遗传力为0.2、0.05,QTL个数和表型性状个数为2进行了广泛的模拟研究。研究结果表明MT-MIM-BC1方法相对MT-MIM方法参数估计的精准度更高。MT-MIM-BC1方法给出了重组率的显性表达式,相对MT-MIM方法能够更精准定位QTL到染色体上。
此外,获得Ω的估计以后,我们可以研究QTL是否显著存在。效应矩阵C的两个假设可表示为
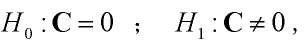
新方法的缺点同一般多区间定位方法一样,当区间数比较多时,计算量非常大。由于基因定位在基因病的研究中起到至关重要作用,特别是有的基因具有多效性,多性状多区间定位方法比多区间定位方法更具有优势。在将来的工作中我们将进一步推广,以便适应更多的标记位点。
猜你喜欢
西北农业学报(2024年7期)2024-07-12 20:26:59
养猪(2022年4期)2022-08-17 07:07:02
中学生数理化·高一版(2019年12期)2019-12-31 06:52:24
当代石油石化(2018年1期)2018-08-10 06:50:54
中国钢铁业(2018年6期)2018-07-26 06:55:00
消费导刊(2017年24期)2018-01-31 01:29:31
辽宁大学学报(哲学社会科学版)(2017年3期)2017-06-21 21:16:59
湖北畜牧兽医(2015年11期)2016-01-11 10:08:24
中学语文(2015年27期)2015-03-01 03:53:28
继续教育研究(2014年2期)2014-02-27 16:10:56
