
粗粒化系统的GROMACS模拟
2018-11-22 03:24:08冯剑
滁州学院学报 2018年5期
冯 剑
1 前言
计算机模拟是继实验和理论研究方法后的又一种科学研究手段受到极大的关注。分子动力学模拟作为重要的工具之一也被广大的化学化工科技工作者所采用。科技工作者常自行编制软件解决自己的研究问题[1]。随着科学研究深入和计算技术的发展,个人编制完整的计算程序用于各类问题求解变得越来越困难。GROMACS[2]、NAMD、LAMMPS等模拟软件已被广泛使用。科技工作者有时常关注研究问题主要影响因素而抛开原子具体细节,理论工作者尤为如此,因而也非常关注粗粒化的模拟工作[3, 4]。
GROMACS特别适用于生物大分子体系的分子动力学模拟,它提供了许多可选的力场,以及很多计算技术,容易扩展到粗粒化模拟领域。知名的MARTINI粗粒化力场[5]就采用了对实际的生物分子中多个原子使用GROMACS的虚拟格点技术进行粗粒化。使用粗粒化模型的GROMACS系统性工作很少报道,本文较完整给出GROMACS粗粒化模拟的一般方法,软件版本是GROMACS v2018.2。
2 力场扩展和体系构建
2.1 扩展OPLS-AA力场
分子动力学模拟必须要选择具体的力场,参数化研究体系。利用GROMACS进行粗粒化处理需要扩展力场的处理能力。为此,需要熟悉相关力场以及GROMACS中力场的组织。本文以OPLS-AA力场为例,对此进行粗粒化处理,其它力场方向相同。
OPLS-AA力场包含多项非键和成键作用,这里仅就其中的非键范德华作用进行说明,更详细的内容可参考相关文献[6]和GROMACS手册。GROMACS处理的非键范德华作用有Lennard-Jones(LJ)和Buckingham两种势能形式,OPLS-AA中使用的是LJ势能函数,其原始形式为
在模拟软件中考虑计算效率,常采用如下变形式
OPLS-AA力场采用后者。不同原子间的相互作用参数根据纯组分值按以下组合规则获得
该组合规则在GROMACS提供的三种规则之一,编号为3。在GROMACS提供的OPLS-AA力场中LJ输入值依然是纯组分的ii和ii。OPLS-AA力场的非键作用规则在力场包含文件forcefield.itp中给与表述,该文件有如下内容:
[ defaults ]
1 3 yes 0.5 0.5
第一列1表示非键范德华作用使用LJ势能,第二列3表示使用编号3的组合规则,yes表示为处于化学键上1-4位原子产生非键作用,后面两列分别为LJ和静电1-4作用的乘积因子。
为了避免直接修改GROMACS库中的原力场造成力场污染,最好的方法是将该力场复制到工作目录。另外,为了与现有的粗粒化力场进行比较,本文使用MARTINI力场的相关参数。修改力场主要有以下几步。
(1)扩展原子类型。打开atomtypes.atp添加几个粗粒化粒子,名称任意但不要与已有的名称冲突。作为示范以下仅添加2个,摩尔质量为72(四个水分子的粗粒化粒子)
CGA 72.00
CGB 72.00
(2)为新的粒子类型添加相互作用。对于非键作用,打开ffnonbonded.itp文件,在最后追加以下几行数据
CGA CGA 72.00 0.0000 A 0 0
CGB CGB 72.00 0.0000 A 0 0
[nonbond_params ]
CGA CGA 1 0.47 5.0
CGB CGB 1 0.47 4.5
CGA CGB 1 0.47 4.0
前面二行分别给定新增两种粒子类型的具体参数,第一列是粒子名称,最后两列是LJ函数的两个参数,这里设置为0。这是因为它们间的所有作用不是由系统按照组合规则生成,而是在[ nonbond_params ]语句块中指定。可以给定这两个参数具体的值。如果希望按照组合规则计算交叉项,则不需要[ nonbond_params ]段。
(3)添加键长作用。打开ffbonded.itp文件,在最后追加以下几行数据,其中第三列1表示谐振势,后面两个参数分别是平衡键长和力常数。
[bondtypes ]
CGA CGA 1 0.47 1250
CGA CGB 1 0.47 1250
CGB CGB 1 0.47 1250
(4)扩展残基数据库。GROMACS的一个强大的功能是利用pdb2gmx模块为生物分子产生拓扑文件。要想使用pdb2gmx为自定义的模型分子产生拓扑就需要扩展残基数据库。新建rtp文件或打开aminoacids.rtp,在最后添加
[ CGA ]
[ atoms ]
CGA CGA 0.000 0
[ CGB ]
[ atoms ]
CGB CGB 0.000 0
如果为多个粒子构成的链状分子生成拓扑,则[atoms]还需要列出分子上所有粒子,同时给出[ bonds ]、[ angles ]和[ dihedrals ]等成键作用项。由于不同的分子均要如此处理,最直接的办法是为每种分子构建一个拓扑包含文件,使用时将该文件包含在系统拓扑文件中。这里给出处理由这两者粒子组成链长为2的分子(残基),成键作用仅有一对键长作用。
[ CGM ]
[ atoms ]
CGA CGA 0.000 0
CGB CGB 0.000 0
[ bonds ]
CGA CGB
2.2 生成系统拓扑和构建分子拓扑
为具体分子构建拓扑首先需要有该分子的结构文件。在GROMACS中常用的有两种格式,一种是pdb格式文件,即蛋白质数据库文件[7],生物大分子的结构一般由该格式提供。还有一种是GROMACS默认的gro文件。对于粗粒化分子,可以使用手工或使用Chem3D等3D分子绘图软件制作。对于单原子分子,使用作图软件插入一个单原子,保存为pdb文件(命名为atom.pdb)并进行适当修改;或使用一个文本编辑器,直接输入相关内容制作atom.pdb文件。该文件仅需2行,如:
ATOM 1 CGA CGA 1 0.000 0.000 0.000
END
注意pdb文件有固定格式,其中7-11列是原子编号,13-16列是原子名称,18-20列是残基名称(不是必须的)。本文还处理双原子分子(命名为mole.pdb),只需要添加一行并做一些改变,内容如:
ATOM 1 CGA CGM 1 0.000 0.000 0.000
ATOM 2 CGB CGM 1 0.000 0.000 0.470
END
这里做了几个改变,首先对分子上的粒子从1进行编号。将分子(残基)命名为CGM,与残基数据库中的[ CGM ]块对应。对第二个粒子z坐标进行修改,距第一个粒子0.47nm。
力场修改完成后就可以为这些分子生成拓扑,执行如下命令即可
gmx pdb2gmx -f atom.pdb -o atom.gro -p topol.top -ff oplsaa -water none
上面命令生成的系统拓扑文件topol.top内容非常简单,其核心内容是
#include "./oplsaa.ff/forcefield.itp"
[moleculetype ]
Other 3
[ atoms ]
1 CGA 1 CGA CGA 1 0 72
[ system ]
Protein
[ molecules ]
Other 1
该系统拓扑文件不仅包含了模拟系统的分子拓扑(前面[ moleculetype ]、[ atoms ]等部分)也包含给系统命名和组成系统的各类分子及其数目等信息(后面[ system]和[molecules]部分)。在GROMACS模拟中,最好为自行制作的分子建立单独的拓扑文件,在使用时添加#include语句包含在系统中。即将最后的[ system]和[molecules]段删除,并删除第一行力场包含文件,将这些内容另存为mol_a.itp,它就是A分子的分子拓扑文件。
[moleculetype ]语句块给定了自行制作分子的名称,该分子系统不能识别,因而给了“Other”名称,它可以修改为任何有意义的名称。第二列3表示不计算连续三个键上的粒子之间的非键作用,粗粒化模拟中该值一般设为1。单粒子分子没有化学键,该项不起作用。修改后为
[moleculetype ]
Mol_A 3
后面的[ molecules ]也要做相应的修改,即
[ molecules ]
Mol_A 1
该行的第二列数据是系统中Mol_A分子的个数,后期需要根据系统的实际情况进行修改。
前面产生的atom.gro结构文件也是具有固定格式的。该文件包含多行内容,其中第一行是任意的说明字符串,第二行是总原子数,最后一行是模拟盒子边长,单位是nm,这些是自由格式。其它各行是原子信息,是固定格式的。这里只有一行,内容如下
1CGA CGA 1 0.000 0.000 0.000
第一列是残基的编号和名称(二项),第二列是原子名称,第三列是原子编号,每项均为5个字符宽度。后面三列是原子坐标,8个字符宽度其中小数位宽度为3。
再次为双粒子分子生成拓扑文件topol2.top:
gmx pdb2gmx -f mole.pdb -o mole.gro -p topol2.top -ff oplsaa -water none
其中该分子拓扑内容如下
[moleculetype ]
Other 3
[ atoms ]
1 CGA 1 CGM CGA 1 0 72
2 CGB 1 CGM CGB 1 0 72
[ bonds ]
1 2 1
同样可以将这部分内容保存为单独的拓扑文件,如moleb.itp,在使用时包含它。在理论研究中的粗粒化模型流体一般都不复杂,可直接根据分子结构依次给出分子中所有粒子([ atoms ]),所有键长作用([ bonds ])和键角作用[ angles ]等。
2.3 构建模拟体系
构建包括不同类型分子的体系是比较复杂的工作,可以借助工具如packmol或自行编程解决。GROMACS也提供了一些模块构建简单系统,如insert-molecules和genconf等。
使用insert-molecules在边长为4.2nm的立方型盒子中插入1000个A粒子,粒子半径为0.47nm。
gmx insert-molecules -ci atom.pdb -o atom_box.gro -box 4.2 4.2 4.2 -nmol 1000 -radius 0.47
再将topol.top中[ molecules ]语句块Mol_A的分子数从1修改为1000。这样就获得模拟体系的结构文件和拓扑文件。
对于双原子分子,构建500个分子的体系可以类似处理,这里给出genconf方法。首先将分子放置一个长方体盒子中并位于盒子中心。
gmx editconf -f mole.pdb -o mole_c.gro -box 0.42 0.42 0.84 -c
将这个盒子在xy维堆积10倍,z维堆积5倍,产生500个分子系统,即
gmx genconf -f mole_c.gro -o mole_box.gro -nbox 10 10 5 -rot
将topol2.top中[ molecules ]语句块Mol_B(原Other修改为Mol_B)的分子数从1修改为500即可。注意对于较长分子链长使用insert-molecules未必能产生足够高的浓度,而genconf则会产生更加有序的系统。
有了系统拓扑和结构文件,就可以按照常规的GROMACS模拟步骤执行模拟,一般有能量最小化、NVT(约束)平衡、NPT(约束)平衡和MD模拟等步骤。本文研究分子不带电,因而在上面各步提供的模拟参数文件(.mdp)中不需要设置静电作用项,rcoulomb和rvdw设置为1.2,tc-grps=System、tcoupl=V-rescale以及pcoup=Parrinello-Rahman等。
3 结果与讨论
图1是使用粗粒化的MARTINI水分子力场参数进行模拟获得298.15K、308.15K至338.15K等五个温度下水的密度,并与美国地质勘探局水科学学院(https://water.usgs.gov/edu/density.html)的水密度数据进行了比较。模拟结果普遍比实验结果大。该力场的创立者也发现这个问题,建议在水中增加少量防冻结粒子,其直径增大到0.57nm[5]。
图1 不同温度下水密度的模拟值与实验值比较
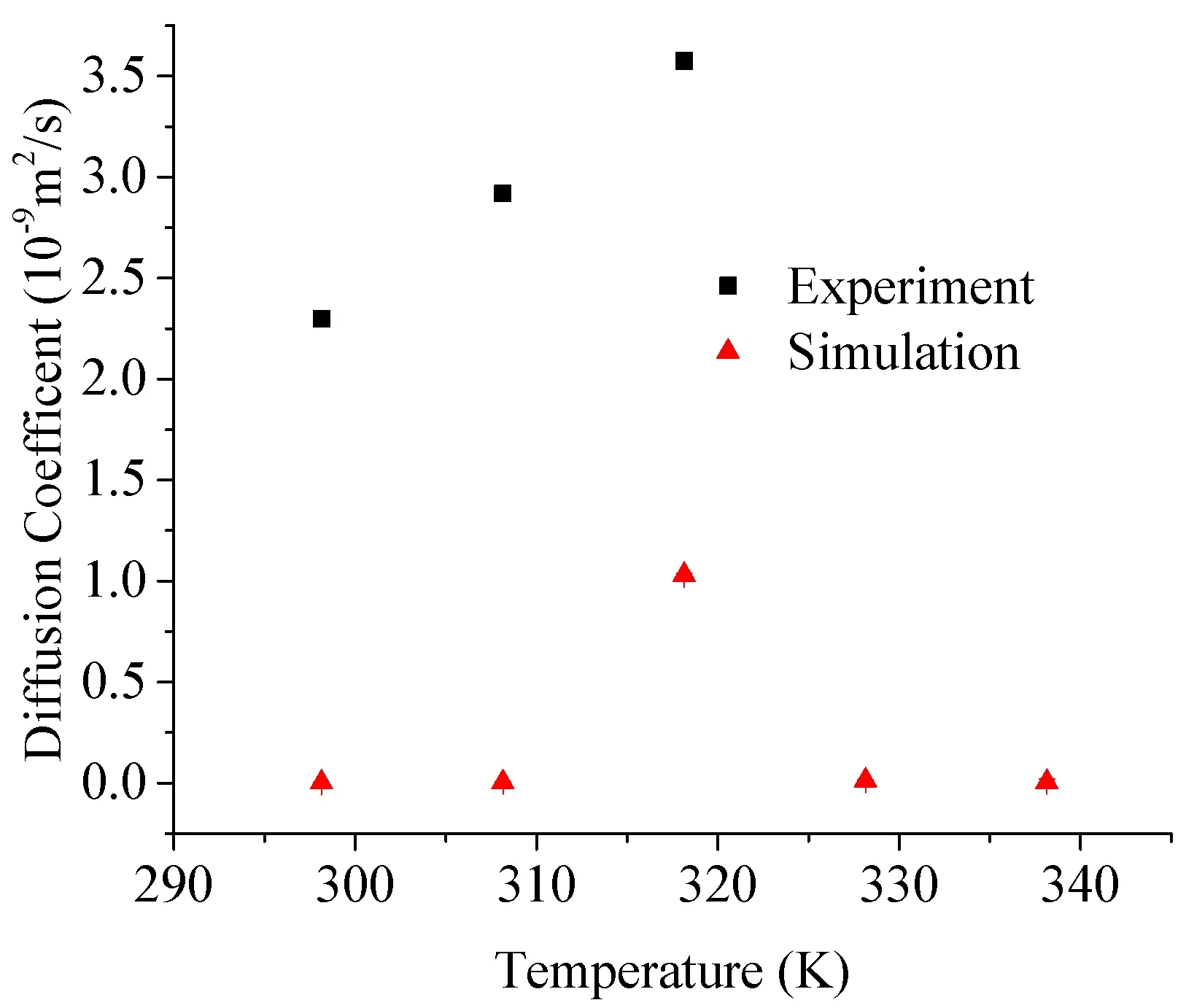
图2 不同温度下水扩散系数的模拟值与实验值比较
MARTINI粗粒化模型获得的密度与实验值有10%左右的误差,而扩散系数差别更大。图2是相应温度下模拟的水扩散系数与实验值的比较[8]。模拟值普遍比实验数据小,除了在318.15K值与实验值数量级一致外,其它温度下有1-2个数量级差别。造成这种现象,一方面是实验水分子和粗粒化的水分子大小和质量不同,需要对模拟值进行标度;另一方面是这个粗粒化的水模型无法精确描述扩散系数之类物理量。为实际体系构筑粗粒化模型本身就是一个艰巨的难题,这些粗粒化的模型往往只能教好描述少数性质,一般无法准确描述众多性质。
图3是两个粒子构成的分子体系各粒子间径向分布函数的模拟。在该模拟中所有粒子间的作用势均设为5.0。相同粒子对CGA-CGA和CGB-CGB间的径向分布函数值在全距离范围内几乎一致。不同粒子对CGA-CGB与它们之间的值大约在小于0.49nm时才表现处明显的差异。从0.472nm始径向分布函数值大于1,这与CGA间CGB存在化学键,键长为0.47nm是一致的。由该图可以得出,尽管在构建该分子体系是有较大的有序性。但经过MD模拟,这种有序被打破,获得了应有的无序状态。

图3 粒子间的径向分布函数
任何MD模拟都有自己的单位集,在GROMACS中长度r单位为1nm,质量单位为原子质量(C质量的1/12),即1u,时间为ps,能量为kJ/mol,力为kJ/mol/nm等。而在粗粒化模拟中质量为m,长度为,时间为,能量为,力为/等。这些量直接设为1,即所谓的约化单位或对比单位。由于导出单位之间彼此关联,一旦选定基础单位后,其它单位就不能再随意变动。因此,在使用GROMACS进行粗粒化模拟时还需要考虑约化单位与GROMACS单位间的对应关系。如将对应于GROMACS中的0.47nm,则由GROMACS得出的长度值只要除以0.47nm,则得到粗粒化系统的长度量。换句话说,将粗粒化单位分别对应于GROMACS中单位的具体值,将GROMACS中获得的相应物理量的值除以其粗粒化单位回代的对应值即可。如上面318.15K模拟的扩散系数为1.0310-9m2/s,扩散系数单位是2/。对应关系—0.47nm、—1ps,则粗粒化模拟的约化扩散系数为4.6610-3。该温度下密度为1051.07kg/m3,粗粒化单位m/3,对应关系为—0.47nm、m—72u,则约化密度为0.91。
4 结论
本文给出了通过GROMACS扩展系统力场,强化软件能力,构建粗粒化体系的方法,并以现有的MARTINI力场参数为例,对两个分子体系进行模拟,获得了满意的结果。同时也介绍了GROMACS体系与用户选择的粗粒化系统间的映射关系。本文工作表明功能强大的GROMACS分子动力学软件具有较大的灵活性和较强的扩展性,可以用于研究更广泛的系统和更复杂的问题。
猜你喜欢
航空材料学报(2023年6期)2023-12-18 05:23:50
小学生学习指导(小军迷联盟)(2023年3期)2023-03-27 09:22:44
中国音乐学(2022年1期)2022-05-05 06:48:46
广西农学报(2019年4期)2019-11-26 11:56:23
物理学报(2018年10期)2018-06-14 06:31:32
上海金属(2015年5期)2015-11-29 01:13:59
上海金属(2015年6期)2015-11-29 01:09:09
数学物理学报(2015年2期)2015-02-28 16:06:39
温州职业技术学院学报(2014年3期)2014-03-11 19:03:38
上海金属(2013年4期)2013-12-20 07:57:07
