
葡萄球菌新型肠毒素rSEP的原核表达与纯化研究
2018-11-16 09:37唐俊妮罗双华赵燕英
西南民族大学学报(自然科学版) 2018年5期
唐俊妮,罗双华,刘 骥,赵燕英,陈 娟,汤 承
(西南民族大学生命科学与技术学院,四川 成都 610041)
金黄色葡萄球菌(Staphylococcus aureus,S.aureus)是一种常见的食源性和医源性致病菌,能够分泌多种毒素,如肠毒素/类肠毒素(staphylococcal enterotoxins/staphylococcal enterotoxins-like,SEs/SEls)、剥脱毒素和中毒性休克毒素(toxic shock syndrome toxin,简写为TSST-1,原命名肠毒素 SEF)等.其中,SEs/SEls的分泌是金黄色葡萄球菌引起食物中毒的主要原因[1-2].SEs/SEls是一类结构相似,具有超抗原(superantigen,SAg)活性的细菌毒素,在极低剂量(ng或pg)时即可刺激T细胞,在T细胞抗原受体上进行增殖,并能明显增强T细胞活性[3-4].SEs/SEls根据发现的基因型及先后顺序目前分为24种,包括传统肠毒素SEA-SEE、新型肠毒素 SEG-SEJ、类肠毒素 SElKSElY等[2].SEs/SEls也可作为超抗原,具有开发成为抗肿瘤药物的潜在可能[5-6].因天然SEs/SEls产量低,从产毒培养的金黄色葡萄球菌上清液中分离纯化SEs/SEls比较困难[7-8].因此,关于上述多数SEs/SEls的活性及功能研究受到限制,大多数新型肠毒素或类肠毒素(SEs/SEls)的致病性及其引起的免疫耐受性等相关作用机制尚不清楚.
葡萄球菌肠毒素P(简称SEP),是新型肠毒素的一种,其分子量约为27.89 kD,具有刺激T细胞增殖及催吐特性的超抗原毒素[9].研究还认为SEP可能与耐甲氧西林金黄色葡萄球菌菌株引起的菌血症相关联[10].如果要对SEP进行深入的结构和功能研究,还需纯化蛋白作为基础.但目前国内外关于SEP蛋白相关报道比较少[9-10].
因此,本研究拟进行sep基因的克隆和重组质粒pET-30a-SEP的构建,在此基础上,进行重组蛋白原核表达条件优化与重组蛋白的纯化,为后续深入研究SEP的结构与功能奠定基础.
1 材料与方法
1.1 实验材料与主要设备
菌株和质粒:表达宿主大肠杆菌(E.coli)菌株DH5α、BL 21(DE 3)、BL 21(DE3)pLySs、Rosetta(DE3)、pMD18-T质粒、原核表达质粒 pET-30a均购自天根生化科技(北京)有限公司.
主要试剂:卡那霉素、氯霉素、IPTG 购自Sigma公司;丙烯酰胺、甲叉丙烯酰胺、咪唑购自Amresco公司;SDS-PAGE上样缓冲液购自康为世纪生物科技有限公司;Ni2+-NTA-Sepharose 购自 GE Healthcar公司;Millipore 10 KD超滤管购自Millipore公司;PCR扩增试剂、核酸分子量标准、限制性内切酶NdeI和XhoI购自宝生物工程(大连)公司.
主要仪器设备:HZQ-F160全温振荡培养箱江苏省太仓市实验设备厂;GHP-9080水式恒温培养箱上海齐欣科学仪器有限公司;MLS-3020电热自动灭菌锅日本SANYO公司;5804R冷冻离心机Eppendorf公司;SC-15数控超级恒温槽、JY92-IIN超声破碎仪宁波新芝生物科技股份有限公司;恒温金属浴、UV-6100分光光度计上海美普达仪器有限公司;伯乐Mini-PROTEAN Tetra Cell电泳仪伯乐科技有限公司.
1.2 实验方法
1.2.1 基因sep的克隆与表达载体构建
金黄色葡萄球菌基因组DNA采用微波加热法提取[11].根据GenBank中报道的金黄色葡萄球菌sep基因序列(NC_004740),采用 Premier 6.0软件进行引物设计,基因 sep克隆引物上游序列(P1):5′-CGTACGCATATGAGTAAAATAAAAAAAACAACA(下划线为NdeI酶切位点,ATG为起始密码子);下游序列(P2) :5′-CGCTAGCTCGAGTCAAGTTGTATATAAATATATATCAA(下划线为XhoI酶切位点);引物由生工生物公司(上海)合成.
PCR 扩增条件:PCR buffer 5 μL,dNTP (2.5 mmol/L) 3 μL,Mg2+(25 mmol/L) 4 μL,上下游引物P1和P2等量混合物 (浓度为10 μmol/L)3 μL,金黄色葡萄球菌基因组DNA模板1μL,Pfu DNA Polymerase 1μL,ddH2O 33 μL,反应体系共计50 μL.PCR反应条件为:95 ℃ 4 min;94 ℃,40 s;65 ℃ 50 s;72 ℃60 s,35个循环;72℃,8 min.扩增所得片段经琼脂糖凝胶电泳检测及PCR产物纯化试剂盒纯化后,与质粒载体pMD18-T进行T-A连接,分别将插入sep基因的pMD18-T质粒与空载质粒pET-30a用NdeI/XhoI双酶切,将上述双酶切获得的纯化片段用T4 DNA连接酶连接,使得目的基因定向克隆至表达载体pET-30a中.将插入sep基因的阳性质粒命名为pET-30a-SEP,同时将质粒进行双酶切验证和送生工生物公司(上海)测序.
1.2.2 最佳表达宿主菌的确定
将酶切鉴定的重组质粒pET-30a-SEP分别转入表达宿主大肠杆菌 BL 21(DE 3)、BL 21(DE3)pLySs和BL 21(Rosetta)中,分别涂布于含相应抗生素的平板上,其中,BL 21(DE 3)和 BL21(DE3)pLysS 涂布于含卡那霉素30 μg/mL的LB琼脂上,BL 21(Rosetta)涂布于含氯霉素10 μg/mL的LB琼脂上,设置对照,37℃恒温培养12~16 h,之后随机挑取含重组质粒的单菌落于含相应抗生素的液体LB中,过夜培养,在设定条件下诱导表达,离心收集菌体,SDS-PAGE分析各表达宿主菌蛋白的表达情况.
1.2.3 重组蛋白不同表达条件优化
选择1.2.2中最优表达菌株,分别确定最优IPTG浓度(37 ℃诱导6 h,IPTG 浓度梯度为0.1、0.2、0.3、0.4、0.5、0.6、0.7、0.8 mmol/L)、最佳诱导时间(37℃,最优 IPTG 浓度,分别诱导 0、2、4、6、8、10、12、24 h)、最佳诱导温度(最优IPTG浓度,最佳诱导时间,分别在37、27、17℃下诱导)和最佳 Zn2+浓度(最佳诱导时间、温度、IPTG浓度,设置Zn2+浓度梯度为0、1、5、10、15、20、25 mmol/L).SDS-PAGE 分析不同条件下蛋白的表达情况.
1.2.4 重组蛋白rSEP纯化
用上述确定的最佳表达条件大量培养重组宿主菌(2 L的培养基),测菌液吸光度(A600nm)至0.6~0.7时,加入最佳浓度IPTG诱导剂诱导,之后离心收集菌体,重悬于Buffer A缓冲液(50 mmol/L Tris-HCl,0.5 mol/L NaCl,pH 8.0)中,冰浴超声破碎(超声3 s,间隔6 s,共30 min),在4℃,12000 rpm 条件下离心20 min收集上清,采用缓冲液平衡好的 Ni2+-NTASEPharose亲和层析柱中反复穿透上清液1 h,随后用洗脱缓冲液(50 mmol/L Tris-HCl,0.5 mol/L NaCl,20~200 mmol/L咪唑,pH 8.0)梯度洗脱,收集目的蛋白,采用SDS-PAGE检测纯化的目的蛋白.
2 结果与分析
2.1 基因sep的PCR扩增及重组表达载体的构建结果
以筛选携带sep基因的金黄色葡萄球菌DNA为模板进行PCR扩增,sep基因扩增的PCR条带见图1(A),凝胶电泳表明 PCR扩增的目的基因片段大小与预期理论大小相符合,证明扩增得到了预期的sep基因片段.进一步对该片段进行克隆和测序鉴定,表明序列正确.通过酶切转化筛选,获得阳性克隆子.进一步对构建的重组表达载体pET-30a-SEP进行双酶切验证,得到了两条与预期大小吻合的酶切产物片段,见图1(B).至此,表明重组表达质粒载体 pET-30a-SEP构建成功.
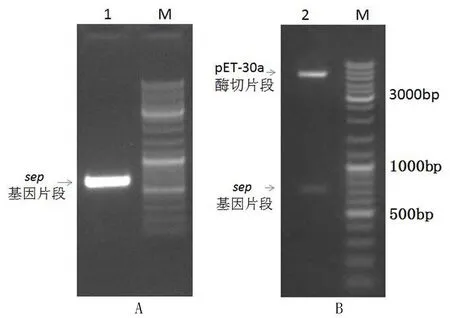
图1 sep基因的PCR扩增电泳图(A)和表达载体pET-30a-SEP双酶切电泳图(B)Fig.1 The results for sep gene PCR amplification and pET-30a-SEP expression vector construction
2.2 原核表达体系不同表达条件的优化结果
针对表达宿主菌的优化,如图2所示,图2A中的表达宿主菌BL 21(DE3)pLySs菌株中融合蛋白表达量明显高于 BL 21(Rosetta)(图 2B)和 BL 21(DE 3)(图2C)中融合蛋白的表达量.因此,后续实验选取BL 21(DE3)pLySs为最佳表达宿主菌株.
针对表达时间的优化,如图3(A)所示,诱导时间为8 h时融合蛋白表达量高(3A中的泳道6);针对IPTG的优化浓度,如图3(B)所示,IPTG浓度为0.2 mmol/L时融合蛋白表达量高(3B中的泳道2);针对温度和Zn2+浓度优化条件,如图3(C)和3(D)所示,结果表明37℃和Zn2+浓度为1 mmol/L时融合蛋白表达量高(3C中的泳道2,3D中的泳道2),从而优化得到重组质粒的最佳表达条件.
进一步在最优表达条件(37℃,0.2 mmol/L IPTG,1 mmol/L Zn2+浓度,诱导 8 h)下,对 BL 21(DE3)pLySs宿主菌的单克隆进行小量诱导和表达验证,如图4所示,多次平行试验均获得了rSEP的高效可溶性表达(图4).证实优化得到的最佳表达条件成功(表达宿主菌选择BL 21(DE3)pLysS、最佳IPTG浓度为0.2 mmol/L、最佳诱导时间为8 h、最佳表达温度为37℃、最佳添加的Zn2+浓度为1 mmol/L).
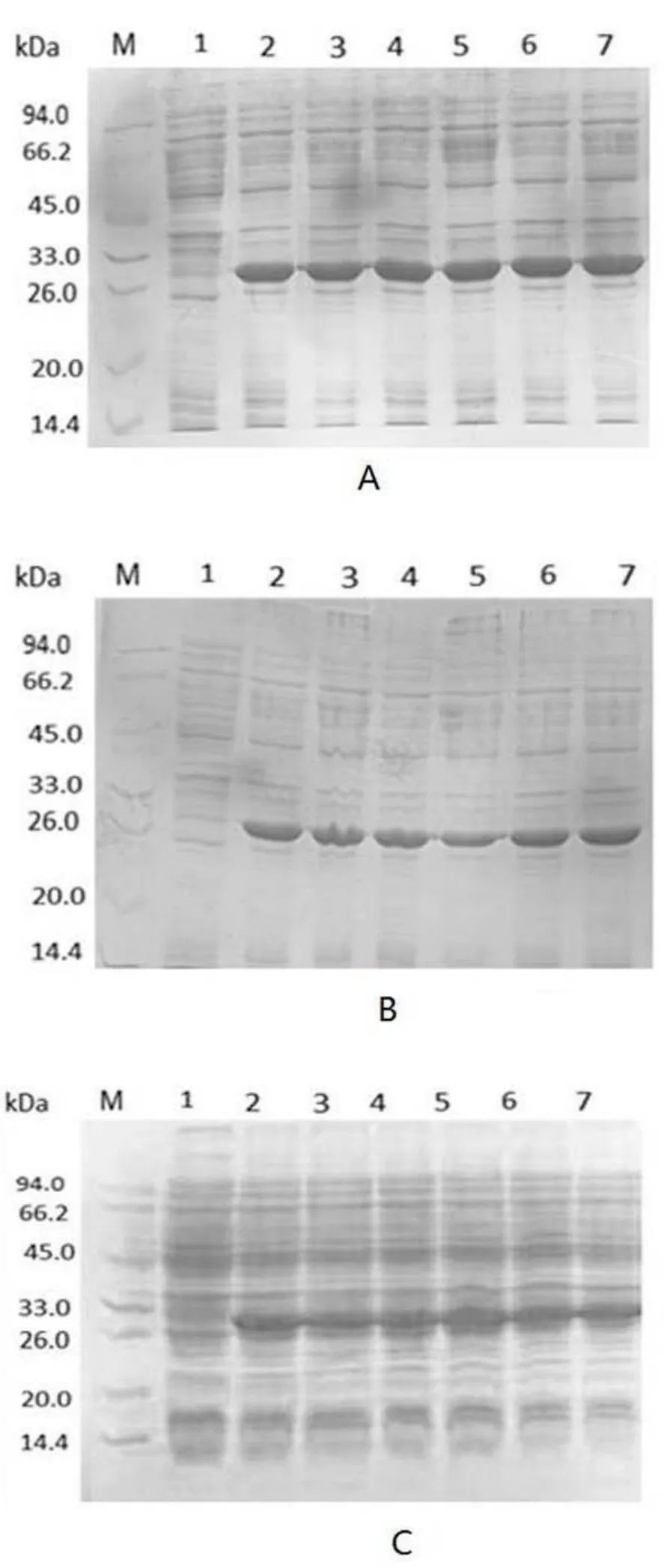
图2 pET-30a-SEP质粒在不同大肠杆菌表达宿主中目的蛋白的SDS-PAGE图Fig.2 The protein expression of pET-30a-SEP in differentE.coli expression host bacteriaA 表达宿主菌为 BL 21(DE3)pLySs;B 表达宿主菌为 BL 21(Rosetta);C 表达宿主菌为 BL 21(DE3);M为蛋白分子量标准,泳道1为未诱导;泳道2-7为6个随机挑取的表达菌株在IPTG诱导下的表达
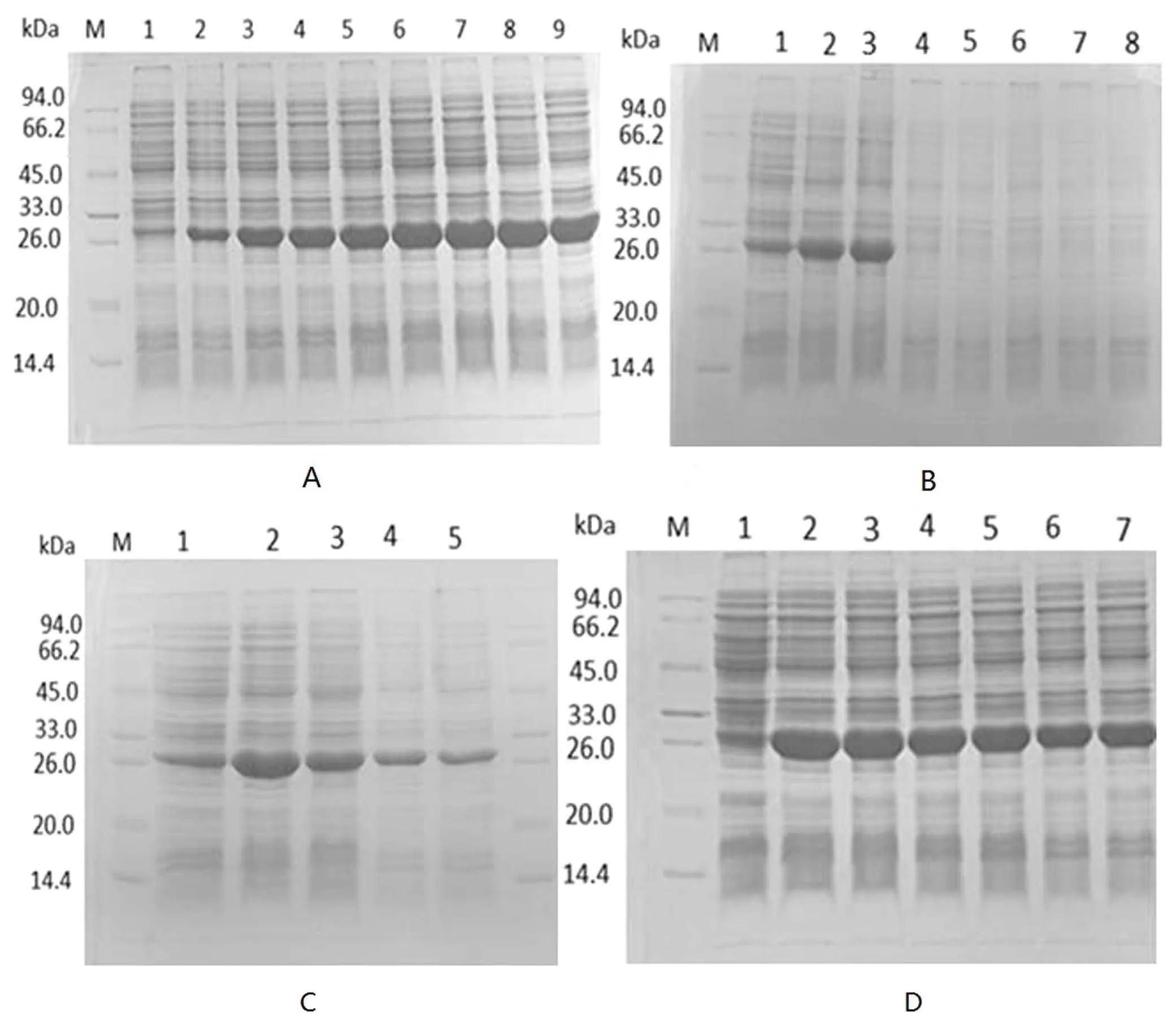
图3 不同表达条件的优化结果Fig.3 The optimization results of pET-30a-SEP protein expression conditions
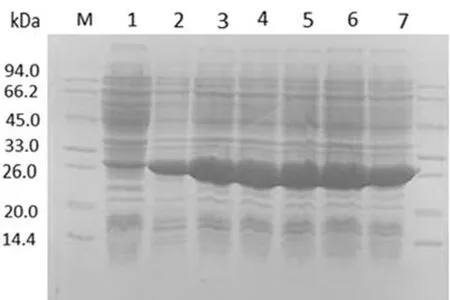
图4 rSEP重组蛋白在最优条件下表达的SDS-PAGE图Fig.4 SDS-PAGE analysis for the optimal expression conditions of rSEP proteinM为蛋白分子量标准;泳道1为未诱导;泳道2~7为最优条件下6个随机挑取的菌株表达的目的蛋白
2.3 重组蛋白rSEP的纯化结果
按上述最优条件进行扩大培养,经过超声波破碎,粗提液进行镍亲和层析纯化,分别用含20、50和200 mM咪唑的缓冲液梯度洗脱杂蛋白,并进行SDSPAGE分析,结果见图5.由图5(A)的第7泳道可见,重组蛋白rSEP采用200 mM咪唑缓冲液洗脱效果最好.进一步验证纯化条件,结果见图5(B),表明获得纯度较高的rSEP蛋白,通过蛋白定量试剂盒测定纯化的rSEP蛋白浓度为1.8 mg/mL,可满足后续研究.

图5 纯化重组蛋白rSEP的SDS-PAGE分析Fig.5 SDS-PAGE analysis for the rSEP fusion protein purification
3 讨论
金黄色葡萄球菌是引起食物中毒的重要致病菌之一,而肠毒素的产生是导致食物中毒的主要原因[12].金黄色葡萄球菌肠毒素基因种类繁多,sep基因作为众多肠毒素基因的一种,在临床上不断被检出,如Hait等[13]在引起食物中毒事件的面包店中检测出sep基因;Liu等[14]从河南省2013~2014年收集的健康动物和医院病人样本中检出sep基因,发现其携带率为14.69%;Wu等[15]从零售蔬菜分离的金黄色葡萄球菌中检出sep基因,携带率高达70.0%.这些研究说明sep基因在金黄色葡萄球菌中普遍流行,该基因表达的SEP蛋白也可能具有潜在的食品安全威胁.但目前关于SEP蛋白的致病机制和功能研究报道不多,面临的主要困难是天然SEP蛋白产量低,从产毒培养液中分离纯化困难,如果需要制备SEP蛋白纯品,需对sep基因编码蛋白进行克隆表达与纯化.因此,本研究中,我们根据GenBank中报道的金黄色葡萄球菌sep基因序列(NC_004740)设计引物,又从我们实验室保存的临床分离金黄色葡萄球菌菌株中筛选获得sep基因序列,为成功构建重组表达载体 pET-30a-SEP奠定了基础.
在表达条件的优化实验中,我们首先对经典的大肠杆菌表达宿主菌 BL 21(DE 3)、BL 21(DE3)pLySs、BL 21(Rosetta)进行原核表达系统的优化,实验发现表达宿主 BL 21(DE3)pLySs效果更佳,BL21(DE3)PlySs是在BL21(DE3)基础上,附带了T7溶菌酶基因,可更严谨控制T7聚合酶的表达.因此,在后续的蛋白表达中,选择BL 21(DE3)pLySs作为rSEP融合蛋白的表达宿主菌.在此基础上,本研究进一步对诱导时间、IPTG浓度、温度、以及Zn2+浓度进行优化,最终获得rSEP融合蛋白的最优表达条件.通过多次验证,均实现了对融合蛋白rSEP的高效可溶性表达.在对表达蛋白纯化的研究中,本研究采用 Ni2+-NTASEPharose亲和层析柱反复穿透上清液,并对洗脱缓冲液中咪唑浓度进行优化探索,针对rSEP融合蛋白,采用200 mM的咪唑洗脱效果更好,最终获得了纯度较高的rSEP蛋白,实现了本研究的目的.
至此,本研究成功构建了金黄色葡萄球菌SEP肠毒素的重组表达载体pET-30a-SEP,对rSEP蛋白进行表达和纯化,并获得了纯度较高的rSEP蛋白,为后续制备单抗、诊断试剂及SEP致病机制研究奠定了基础.同时,本研究的优化条件也可为其他葡萄球菌肠毒素蛋白的表达与纯化提供借鉴.
猜你喜欢
食品安全导刊(2021年20期)2021-08-30
猪业科学(2021年3期)2021-05-21
数学大王·低年级(2020年8期)2020-08-14
中国医药指南(2019年14期)2019-01-07
创新作文(1-2年级)(2018年2期)2018-09-13
创新作文(小学版)(2018年4期)2018-07-06
学习报·教育研究(2017年33期)2017-08-31
农家科技下旬刊(2017年3期)2017-04-26
小雪花·初中高分作文(2016年9期)2016-05-14
中国医药生物技术(2015年4期)2015-12-26
