
从制药企业角度谈完善我国仿制药一致性评价体系
2018-11-14 03:07娜赵鸿剑梁
机电信息 2018年32期
周 娜赵鸿剑梁 毅
(1.中国药科大学,江苏南京211198;2.苏州市工业园区食品药品安全稽查大队,江苏苏州215000)
0 引言
我国是一个以生产仿制药为主的国家,国内生产的药品中97%是仿制药。与原研药相比,仿制药具有较好的经济效益和社会效益。
但不可否认,我国仿制药与世界先进的仿制药还存在以下四个方面的差距:一是原料的差距,二是新技术的差距,三是辅助材料的差距,四是创新剂型的差距。开展仿制药质量和疗效一致性评价工作,全面提高仿制药质量是《国家药品安全“十二五规划”》的重要任务,是持续提高药品质量的重要手段。
1 我国仿制药质量一致性评价概况
1.1 政策依托及实施现状
仿制药(Generic drug)是指以原研药(Reference listed drug)为参考对比,在剂量、安全性、效力、质量、作用以及适应证上相同的一种仿制品。仿制药一致性评价旨在通过质量一致性评价淘汰内在质量和临床疗效都不达标的品种,促进我国仿制药水平的提高,达到或接近先进国家水平。
按照《国家药品安全“十二五”规划》要求,国内相关机构已经开始了仿制药质量一致性评价工作。为推进这一工作,国家食品药品监督管理局于2013年2月发布了《关于开展仿制药质量一致性评价工作的通知》,拟定分批对2007年修订的《药品注册管理办法》实施前批准的基本药物和临床常用仿制药进行质量一致性评价。
2013年12月,中国药品检定所发布关于征求《口服固体制剂参比制剂确立原则(草案)》意见的通知。
同年,药检所发布征求《普通口服固体制剂溶出曲线测定与比较指导原则(草案)》意见的通知。其后,又颁布了美托洛尔、盐酸氨溴索等5个品种的溶出曲线一致性评价方法。2015年8月18日,随着国务院《关于改革药品医疗器械审评审批制度意见》的发布,仿制药一致性评价又重新得到重视,与化学药品注册分类改革并驱。
1.2 实施流程
图1为企业开展仿制药质量一致性评价的整体流程。我国制药企业在开展仿制药质量一致性评价工作的过程中面临着以下几方面的挑战:
1.2.1 参比制剂
参比制剂首选国内上市的原研药品,参比制剂由企业自行购买,在境外购买用于仿制药质量和疗效一致性评价研究用的参比制剂,需要办理一次性进口药品批准文件。
1.2.2 药学一致性评价
仿制药质量一致性评价的指标及原研药的关键质量属性难以确定,仿制药药学质量是否需要与原研药完全一致也有待确认。
1.2.3 生物等效性实验(bioequivalency,BE)
生物等效性实验缺乏相关的法律法规依据,如何进行完整的生物等效性实验也有待研究。
2 我国仿制药质量控制及其质量一致性评价所存问题
2.1 技术层面
2.1.1 原料、辅料、处方环节
原辅料控制、处方、工艺、生产等环节是我国制药业的薄弱环节。生产过程中物料的选择对最终产品的质量有着重大影响。
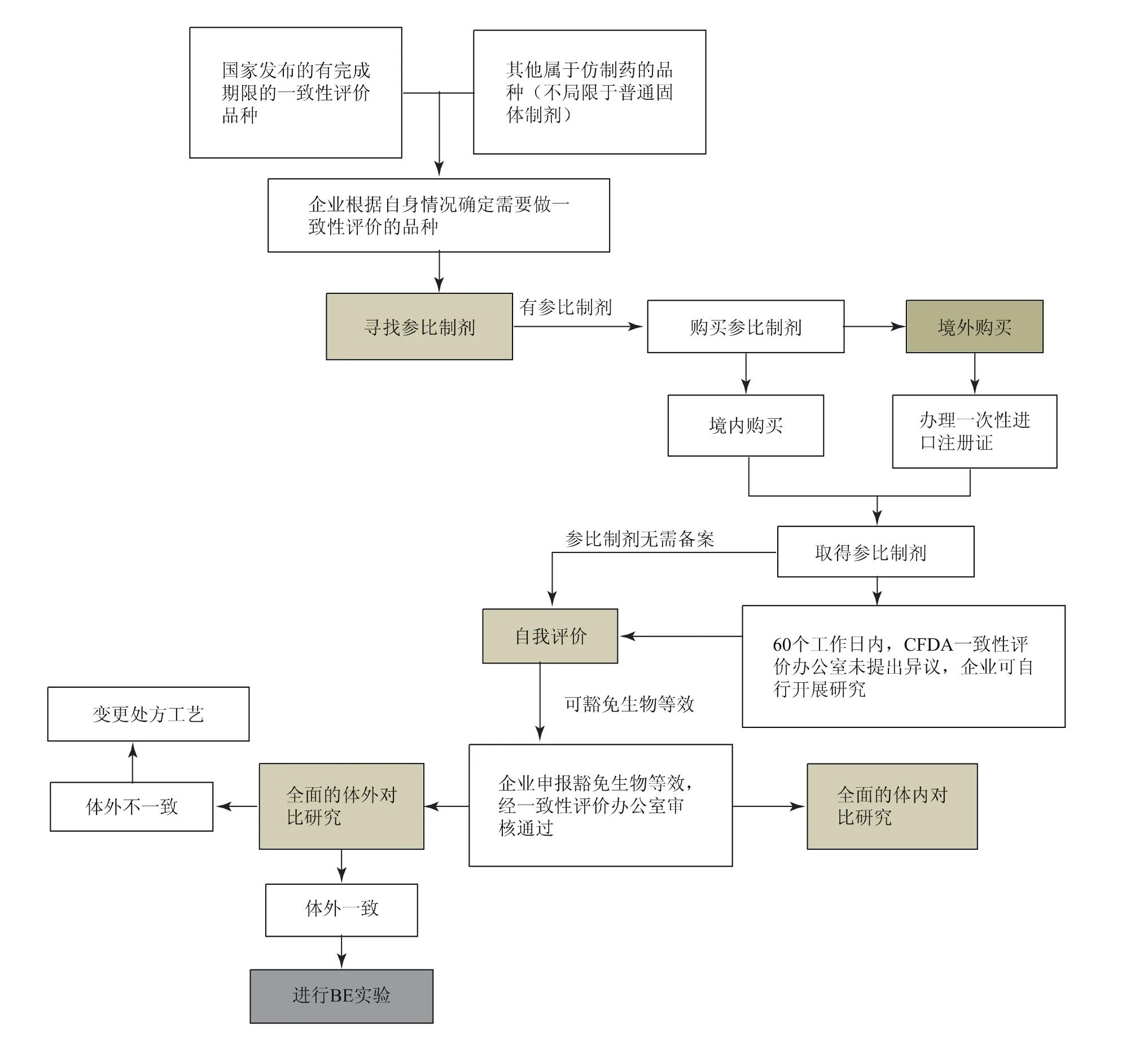
图1 企业开展仿制药质量一致性评价的整体流程
仿制药和原研药在原料、辅料、处方、工艺等方面均不同,不同厂家生产的物料在质量上也会存在差异。在生产过程中,涉及的物料通常有很多种,不同的物料有不同的质量差异,最终集合在最终产品中,将对产品质量造成重大影响。
仿制药在处方的选择上也会与原研药存在差异,主药与辅药之间相容性差,则药品在贮存过程中会产生原研药质量标准中未予以规定的杂质。
2.1.2 工艺环节
仿制药是与原研药具有相同的活性成分,在剂量、剂型、给药途径、安全性和有效性、质量、治疗作用以及适应证上没有明显差异的仿制品。生产工艺是影响药品的物质基础,产品处方相同而工艺不同,所得的物质基础就不同,其安全性、有效性就可能不同。
一般原研药由于保密的原因,其工艺具体参数、条件并未具体公开。制药工艺是根据原研药的质量标准,反向倒推而得到,且只能保证主要成分的质量标准是一致的。不可否认,不同的工艺所得的产物肯定不同,我们无法保证仿制药和原研药是完全相同的。
2.1.3 生产过程的严谨性
单纯依靠抽检是无法保证药品全部合格的。在建立仿制药与原研药一致性指标时,应将药品生产的过程控制纳入仿制药一致性评价体系中。
美国食品药品监督管理局(FDA)的《橙皮书》上明确规定,仿制药的生产严格按照动态药品生产质量管理规范(Current Good Manufacture Practices,cGMP)的要求,即仿制药在生产过程中严格按照标准程序进行生产,按照规定的中间控制标准进行检验,在药品生产全过程中保证药品的质量。
2.2 企业层面
在仿制药一致性评价工作中,制药企业居于主导地位,理论上,仿制药疗效如果接近或者等同于原研药,将会获得更高的市场认同。药品的一致性评价相当于重新研发,需要耗费大量资源。企业需要从辅料的选择、工艺的稳定性等多方面保证产品的质量。除此之外,仿制药质量一致性评价,从选择参比制剂、开展生物等效性实验、一致性评价结果,到现有政策环境支持和技术要求,都需要面对挑战,太多政策的掣肘及不确定因素让企业心有余而力不足。
2.3 方法层面
2.3.1 评价方法处于摸索阶段
仿制药质量一致性评价是一项复杂的工作,其涉及处方、工艺、原辅料、储运和包装等药品生产的整个过程。仿制药与原研药质量一致,不仅体现在体外检验指标(包括有关物质、溶剂残留、含量、溶出度或释放度等)一致,还体现在体内安全性和有效性方面的一致性。物质基础的一致性主要通过处方、质量标准、晶型、粒度和杂质等主要药学指标来确认,临床疗效的一致性主要通过一些体内外的实验来评估。
溶出曲线和生物等效性实验(BE)均是一致性评价的标准。现有实验证明,多个生物等效的药物,其体外溶出曲线是有较大区别的,即使是原研药物,也会出现不同批次间溶出曲线不一致的现象。特别是对一些缓控释或其他释放机制的制剂而言,还需要考虑其他因素。
溶出度是考察普通口服制剂和缓控释口服制剂在胃肠道中释放速度和程度的参数。但在实际情况中,某些特定人群,特别是胃酸缺乏者、联合用药者和年老体弱者,其胃肠道环境、蠕动等与健康者有很大的不同。
我国仿制药质量一致性评价的主要对象为2007年《药品注册管理办法》出台之前已上市的药品,涉及面广。因此,采用临床验证的方法进行一致性评价存在很大困难。一般情况下,采用能灵敏反映体内疗效的体外实验是优先考虑的评价方法。针对不同剂型,评价方法的选择也存在不同。对于口服固体制剂,我国借鉴了日本的做法,采用溶出度实验作为质量一致性评价的手段。对于注射剂,由于不存在药物吸收问题,主要关注安全性指标,对主药通过完善质量标准来进行评价;对于其他剂型,将结合剂型特点,设定合理的评价方法和标准。
2.3.2 参比制剂的选择
参比制剂是指用于仿制药质量和疗效一致性评价的对照药品,通常为被仿制的对象,如原研药品或国际公认的同种药物。参比制剂应为处方工艺合理、质量稳定、疗效确切的药品。
在美国,参比制剂的确立是以法律的形式写进《橙皮书》。在欧盟,参比制剂为成员国内已经上市8年以上的具体品种,参比制剂的选择也以法律的形式规定。
从我国国家食品药品监督管理局发布的《关于仿制药质量和疗效一致性评价参比制剂备案与推荐程序公告》可以知道,目前我国参比制剂目录尚未建立完全,参比制剂的获得途径为“自主申请—专家评估—行政批准”。无明确法律规定参比制剂的获得途径,从一致性评价开始到结束,参比制剂需要不停地申请备案,对企业和监管部门造成极大的负担,严重阻碍仿制药质量一致性评价工作的进展。
2.3.3 受试者的局限性
生物等效性实验是评价仿制药质量一致性的“黄金标准”。除麻醉药、I类精神病药、高毒性药物、妇儿用药等特殊药物以外,一般药物的生物等效性实验都选择18~40岁的健康男性受试者。即使通过实验得出两种药物生物等效,也只能说明两者在健康男性体内的吸收速度和吸收程度没有显著的差异,并不能保证两者在所有人群中都有相同的疗效。
3 完善我国仿制药质量一致性评价体系
3.1 加强制药企业的药品生产全过程管理
我国制药行业的研究基础薄弱,部分企业不重视GMP的执行,对原辅材料来源的审计、原料晶型的控制、处方及工艺参数的筛选、生产过程及最终产品的控制不够严格。随意变更处方,改变工艺流程,偷工减料,这些都直接影响了药品质量和产品稳定性。
3.2 政府部门为制药企业营造良好的政策环境
目前,我国国家食品药品监督管理局向社会公布了已通过一致性评价的药品品种,药品生产企业可在药品说明书、标签中予以标注。除此之外,在医保方面予以适当支持,医疗机构应优先采购并在临床中优先选用。
参比制剂的获得是制约企业进行仿制药一致性评价的一个关键因素。一般情况下,原研药一般是国外的产品,如果该原研药未在国内上市,参比制剂需要到国外购买,涉及一次性进口药品的审批,需要提供质量标准和检测报告,且现行一次性进口批件有效期短,并不能满足需要进口多批次参比制剂进行一致性评价的需求。因此,原研药的审批流程,应适当放宽标准,以满足企业开展仿制药质量一致性评价的需要。
3.3 积极发挥企业在一致性评价工作中的主体作用
仿制药一致性评价涉及到企业已有的批准文号,还涉及到技术更新、知识储备等企业方方面面的改革。企业需发挥自身的能力,并联合其他企业,参与到一致性评价当中。政府应制定相关法规明确要求已上市的原研企业积极配合我国仿制药一致性评价工作,在参比制剂的制定、质量标准的研究、溶出曲线的制定等方面相互配合。
4 结语
政府和企业应明确各自在仿制药质量一致性评价中的作用。企业是仿制药质量一致性评价工作中的主体,是仿制药一致性评价工作的具体实施者。政府部门作为指导者、服务者和协调者,在不违反重大方针政策的前提下,应尽己所能,为企业开展仿制药一致性评价提供绿色通道,协调各方面工作。
猜你喜欢
公民与法治(2022年5期)2022-07-29
中国医院用药评价与分析(2022年5期)2022-06-23
家庭医药·快乐养生(2022年6期)2022-06-23
教学考试(高考物理)(2021年5期)2021-11-08
中医眼耳鼻喉杂志(2021年1期)2021-07-22
今日农业(2020年18期)2020-12-14
大众健康(2019年6期)2019-06-10
中成药(2017年4期)2017-05-17
燕山大学学报(2015年4期)2015-12-25
