
RACK1依赖的miRNA途径参与调节植物叶绿素的生物合成
2018-11-09 08:32上官燕李娇爱孙亿敬刘祉辛孙旭武
上海师范大学学报·自然科学版 2018年5期
上官燕, 李娇爱, 王 哲, 孙亿敬, 刘祉辛, 孙旭武
(上海师范大学 生命与环境科学学院 植物种质资源开发协同创新中心,上海 200234)
0 引 言
microRNA(miRNA)是21~24个核苷酸的RNA非蛋白质编码基因的产物[1].在植物和动物的基因组中含有大量miRNA编码基因,它们首先被转录成较长的primary microRNAs(pri-miRNA),随后在RNA酶Ⅲ样DICER酶的作用下从pri-miRNA中释放出长度大约为20~22 nt的miRNA.成熟的miRNAs进一步被整合到ARGONAUTE(AGO)效应蛋白中以形成一个RNA-induced silencing complex(RISC),RISC的主要调节目标是转录后的信使RNA(mRNA)[1].miRNA介导的基因表达控制参与调节了植物生长发育的诸多方面,包括根和叶的发育、激素应答、发育转变和应激反应[1].
在模式植物拟南芥(Arabidopsisthaliana)中,4种DICER-like(DCL)蛋白之一的DCL1主要参与miRNA的产生[2-4].蛋白DAWDLE(DDL),TOUGH(TGH),HYPONASTIC LEAVES 1(HYL1)和SERRATE(SE)与DCL1相互作用来精确和有效地调节miRNA的产生[5-12].在miRNA产生期间,长的pri-miRNA转录物被进一步切割形成precursor miRNA(pre-miRNA),并从其中释放出miRNA双链体.然后,miRNA通过HUA增强子1(HEN1)被甲基化,并且主要与拟南芥中10种AGO蛋白之一的AGO1结合[11,13-14].HEAT SHOCK PROTEIN 90(HSP90)和cyclophilin 40蛋白SQUINT(SQN)与AGO1相互作用并确保有效的miRNA加载[15-17].AGO1去除miRNA双链体的passenger strand(或miRNA *),允许miRNA guide strand识别靶RNA内的互补序列区域[1,18].AGO1与靶序列的结合导致miRNA结合位点内的mRNA切割,或在内质网上进行的翻译被抑制.
除了miRNA,植物还富含另一种类型的small interfering RNA(siRNA),其与miRNA的结构、基因组和功能均相似.siRNA源自转基因的模板,以及内源性重复序列或转座子[19-21].在模板、重复序列及转座子方面,miRNA与来自转基因的siRNA的一个关键区别是miRNA的靶基因不是产生miRNA的基因,而siRNA是针对生成它们的序列的.最近,一种新的与miRNA相似的siRNA,被命名为trans-acting siRNA(ta-siRNA)[22-23].ta-siRNAs起源于产生非编码模板的基因座,它们是miRNA的靶基因[24].由miRNA介导的转录本的剪切招募一个依赖RNA的RNA聚合酶 (RdRP),将剪切的转录本作为模板,以形成长的双链RNA(dsRNAs),它们是多个ta-siRNAs的来源.miRNA介导的pre-RNA的剪切对ta-siRNAs的形成至关重要[25].
SPETH等[26]利用酵母双杂交筛选的策略,鉴定到了一个与SE互作的蛋白receptor of activated C kinase 1 (RACK1).进一步分析表明RACK1通过和SE互作,作为AGO1复合物的一部分,参与调节一些pri-miRNA的积累和加工.rack1突变体导致一些miRNA的靶基因的miRNA的过量富集,并改变了植物对脱落酸(abscisic acid,ABA)的响应和叶序的发育[26].RACK1存在于几乎所有的真核生物中,具有7个WD40-b-propellers,其可以与多种蛋白质相互作用[27-29].RACK1本身不具有酶活性,而是充当蛋白质之间相互作用的桥梁或与其他蛋白质竞争结合位点,从而抑制特定的相互作用.RACK1通常在蛋白质组学方法中被鉴定,表明RACK1是许多复合物的一部分,其中,它控制复合物的形成和稳定性,或者为调节子创建对接位点[30].直接与RACK1相互作用的几种类型的蛋白已经得到了描述[27].RACK1可以作为真核生物40S核糖体亚基的核心成分,能直接调节翻译来响应各种刺激[27,29].在细胞核中,RACK1作为衔接子连接激酶与其底物,并调节转录[31-34].
在动物中研究发现,RACK1在miRNA途径中起作用.秀丽隐杆线虫(Caenorhabditiselegans)的AGO蛋白ALG-1直接与核糖体相关的RACK1结合,意味着秀丽隐杆线虫RACK 1可能是被翻译的mRNA上的ALG-1的锚点[35].在另一项使用肝肿瘤细胞系的研究中发现,RACK1影响miRNA效应复合物中的加载,并且参与调节KH-type splicing regulatory protein(KSRP)的定位,KSRP参于促进一小部分pre-miRNA的成熟[36].虽然在秀丽隐杆线虫中发现RACK1的敲低会导致更高的miRNA水平,而在人类细胞中的结果则是相反的[35-36].这些看似矛盾的结果和发现暗示,在许多不同的细胞过程中,RACK1在miRNA途径中更具体的功能可能被其多效性作用所遮蔽.为了进一步探索RACK1在植物miRNA途径中的潜在功能,本文作者对美国国家生物技术信息中心(National Center for Biotechnology Information,NCBI)上共享的有关rack1突变体的small RNA-sequence的数据进行了系统的生物信息学分析,发现rack1突变体显著影响着27,24,及21 nt的small RNA的富集.分析差异small RNA以及其靶基因发现,rack1突变体主要影响了细胞核组分、叶绿体以及线粒体组分相关的基因的表达.
1 材料与方法
从NCBI网站下载small RNA-sequence数据包GSE40579(https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE40579),根据规范的生物信息学分析流程进行数据分析.采用Illumina Hiseq 系统测序所得的数据,称为 raw reads 或 raw data.使用cutadapt软件去掉接头序列,并按长度对序列进行过滤,去掉长度小于15 bp及大于41 bp的序列.使用Fastx_Toolkit软件,对序列进行Q20质控,保留Q20达到80%及以上的序列.使用NGS QC Toolkit过滤掉含有N碱基的reads,最终得到高质量的clean reads并用于后续分析.使用Fastx_Toolkit (version 0.0.13)对clean reads中的重复序列进行去冗余,得到uniq reads.使用自行编写的程序对clean reads进行长度分布统计,对uniq reads的重复序列进行统计.
Clean reads 将和指定物种基因组进行比对,并统计比对率.由于小RNA切胶范围为 18~35 bp,miRNA长度分布主要集中在21~22 bp,piRNA主要集中在30 bp左右,transcription initiation RNA(tiRNA)以及tRNA-derived fragments(TRFs)主要集中在30~35 bp.Small RNA测序reads中包含:miRNA、Piwi-interacting RNA(piRNA)、tRNA、rRNA、snoRNA等,得到clean reads后,使用bowtie或者blast软件与上述small RNA 的数据比对,进行small RNA的分类注释.标准流程中会对鉴定出来的miRNA进行差异筛选、靶基因预测,以及功能富集分析.通过分类注释,没有注释的reads即unannotation 序列,将会使用mirdeep2 软件进行novel miRNA 预测.
2 结 果
2.1 rack1突变体的small RNA sequence数据的统计分析
分析rack1突变体和野生型 (WT)的clean reads的长度分布,结果如图1所示.由图1可知,rack1突变体中的不同的small RNA的分布发生了改变.

图1 clean reads 的长度分布统计.(a) rack1突变体样本第一组;(b) rack1突变体样本第二组;(c) WT样本第一组;(d) WT样本第二组
Clean reads 的长度分布有助于比较不同处理样本的小 RNA 情况,一般情况下样本中的长度分布峰值为21~25 bp,动物中的峰值有时为31~33 bp,可能为piRNA或tRNA.将clean reads依次和Rfam数据库、cDNA 序列、物种重复序列库、miRBase 数据库等进行对比注释.首先将clean reads与参考基因组进行比对,统计比对上的参考基因组上的reads数比例,以及未比对上的参考基因组的比例.使用blastn 软件,对clean reads序列同Rfam (version 10.0) 数据库进行比对,提取 E-value 小于等于0.01 的结果,注释出rRNA、snRNA、snoRNA和tRNA等序列.尽可能地发现并去除其中可能的rRNA、scRNA、snoRNA、snRNA和tRNA,并进行small RNA的去除.最终将这些注释到Rfam数据库中的序列进行过滤去除,不用于后续的已知miRNA比对以及新的miRNA预测.使用bowtie软件,将过滤后的Rfam数据库reads同转录本序列进行无错配比对,能完全匹配上转录本序列,并且长度大于26 bp的序列,认为可能是mRNA的降解片段序列,长度小于15 bp的序列不用于后续miRNA的预测,过滤去除这些序列.最终提取出比对结果中序列长度为15~26 bp的序列,将之与无法比对上转录本的序列进行合并,用于后续的已知miRNA比对,及新的miRNA预测分析.重复上述步骤,使用Bowtie软件将过滤后的reads与miRBase中该miRNA的成熟体序列进行无错配比对,比对上的序列被认为是已知的miRNA,并作为miRNA的表达量统计,进行后续差异分析.
miRNA由前体发育为成熟体的过程是由Dicer酶切完成的,一般来说,酶切位点的特异性使得miRNA成熟体序列首位碱基对于碱基U具有很强的偏向性.其他的位点也有统计,如:2~4 号位一般缺少U,第10号位偏向于A(第10号位一般是miRNA对其靶基因发生剪切作用时的剪切位点).对rack1突变体和野生型样本中的已知miRNA碱基偏好性进行了统计分析(图2),结果表明rack1突变体和野生型样本之间的miRNA碱基偏好性没有显著差异.通过上述各数据库的比对注释,最终汇总出各样本中的small RNA分布情况(图3).

图2 各碱基偏好性统计.(a) rack1突变体样本第一组;(b) rack1突变体样本第二组;(c) WT样本第一组;(d) WT样本第二组(Position 代表在smallRNA中的碱基位置,顺序从5′~3′)

图3 Reads 分类注释饼状图.(a) rack1突变体样本第一组;(b) rack1突变体样本第二组; (c) WT样本第一组;(d) WT样本第二组
2.2 新miRNA的预测
将small RNA测序数据和不同的数据库进行了比对,去除tRNA、snRNA、rRNA 等small RNA,将过滤后的序列和miRBase数据库比对,统计已知miRNA.使用Mirdeep2软件对未注释上的序列进行新的miRNA预测.采用RNAfold软件,对能够比对上基因组的序列进行二级结构预测,对能够形成miRNA发夹前体的序列,归类为可能的新 miRNA序列.提取出预测的miRNA的mature序列和star序列,同时进行novel miRNA的定量分析.对预测得到的miRNA的结构信息、序列信息,以及样本归属情况进行生物信息学分析,包括:miRNA hairpin发卡结构(二级结构)的预测结果,clean miRNA reads比对参考基因组的位置,错配情况,样本归属情况等.定位预测已知miRNA在hairpin 中的具体定位(比对结果)以及每条已知miRNA前体与成熟体的read比对情况及错配统计.预测到了17个novel miRNA.图4为所关注的叶绿体的chloroplast_35062以及在rack1突变体中显著上调的2_12894的结构和错配信息.其中,sample 列表示 sample 缩写标签.reads 列表示比对在该处的 read 数目,mm 列表示错配碱基个数(错配碱基在比对图中用大写字符表示).在比对区域内参考序列中彩色碱基区域为测序片段所覆盖到的区域.
2.3 rack1突变体的miRNA的表达水平分析
统计了已知miRNA和新预测出的miRNA序列在rack1突变体和野生型植物中的表达水平,如图5(a)所示,miRNA表达量计算采用5个统计量:最小值、第一四分位数(25 %)、中位数(50 %)、第三四分位数(75 %)和最大值,来粗略地检测数据是否具有对称性以及分布的分散程度等信息.进一步对在rack1突变体和野生型植物中的miRNA表达水平作了transcript per million(TPM)密度分布分析,如图5(b)所示,检测各个样品的基因表达模式,不同颜色的曲线代表不同的样本,曲线上点的横坐标表示对应样品TPM的对数值,点的纵坐标表示概率密度.对rack1突变体和野生型植物中的miRNA 表达水平进行了相关性检验,结果如图5(c)所示,验证了实验的可靠性和样本选择的合理性.对rack1突变体和野生型植物中的miRNA 表达水平作了主成分分析(principal component analysis,PCA),如图5(d)所示,WT 和RACK 1的miRNA的分布发生了较大的差异.
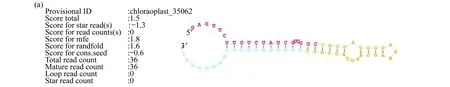
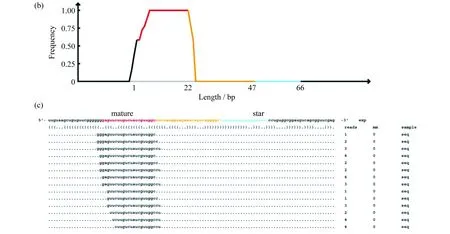
图4 预测得到的新miRNA 结构图.(a) 为miRDeep2软件对miRNA前体的打分和比对到成熟序列、环状结构、star序列上的reads数及其前体的二级结构预测图(红色为成熟序列,黄色为环状结构,紫色为star序列);(b) 显示比对到本条前体上的reads分布;(c) 包含了成熟序列、环状结构、star序列的位置(紫色为miRDeep2软件预测的star序列,亮蓝色为测序reads支持的star序列)
2.4 rack1突变体的miRNA的差异表达分析
对rack1突变体和野生型植物中的miRNA表达水平进行了差异表达分析,在得到差异表达之后,对差异表达miRNA进行靶基因预测及gene ontology (GO)功能显著性和KEGG pathway显著性分析.随后对差异表达miRNA进行非监督层次聚类分析,结果如图6所示.计算多个样品两两之间的距离,构成距离矩阵,合并距离最近的两类为一新类,计算新类与当前各类的距离,循环以上步骤直至只有一类为止.用挑选的差异miRNA的表达情况来计算样品之间的相关性,一般来说,同一类样品能通过聚类出现在同一个簇中,聚在同一个簇中的 miRNA 可能具有相似的生物学功能.
2.5 rack1突变体的差异 miRNA 靶基因预测及靶基因功能分析
植物miRNA与靶mRNA几乎完全互补配对,而大多数动物miRNA与其靶mRNA 不是完全互补配对的.植物miRNA可以与靶mRNA的任何区域结合(主要是蛋白编码区),通过切割靶mRNA或抑制靶mRNA翻译实现对基因表达的调控,动物miRNA主要针对靶mRNA的3′非编码区域(3′UTR),作用机制为抑制翻译.针对差异表达分析得到的miRNA,使用Target Finder软件进行植物的靶基因预测.对预测的靶基因进行GO富集分析,如图7(a)所示.进一步对预测的靶基因作了KEGG pathway功能分析,结果如图7(b)所示,每个小点对应一个通路,p值越大,表示该通路中基因个数越多.由图7可知,RACK1调控的miRNA的靶基因主要参与调节防御反应、盐胁迫、钙离子代谢、叶绿体功能、叶绿素合成,以及核基因表达等.
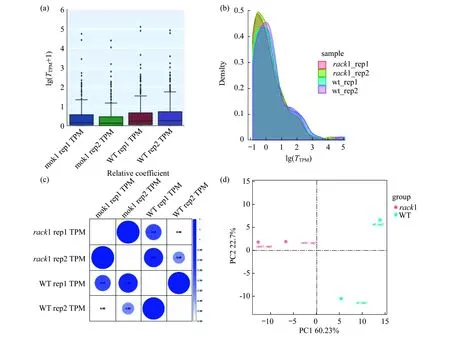
图5 miRNA 表达分析.(a) 样本箱线图;(b) 样本密度图 (density指概率密度);(c) 样品间相关系数热图;(d) 样本 PCA 结果
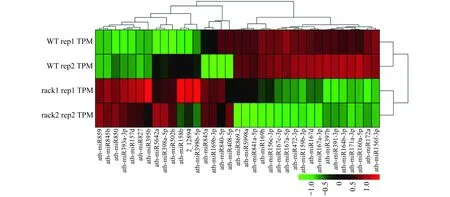
图6 差异miRNA 热图
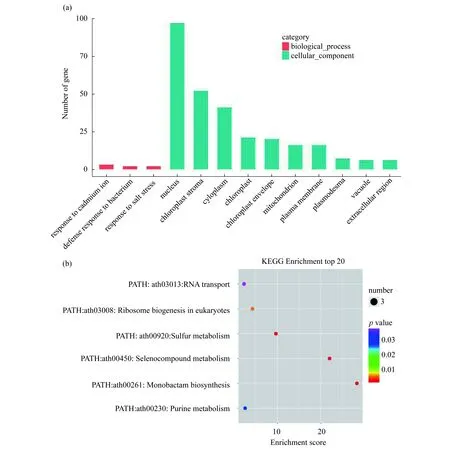
图7 预测miRNA 靶基因的功能通路分析.(a) GO 富集 top 10 条目图;(b) KEGG 富集 top 20 气泡图
3 讨 论
对rack1突变体和野生型拟南芥的small RNA-sequence数据进行了重新分析,并对分析结果进行了深入的整理和挖掘.结果表明:rack1突变体主要影响了27 nt和24 nt的small RNA产生.鉴定到了17个novel miRNA,其中2-12814的表达在rack1突变体中发生了显著性差异上调,其靶基因主要是一系列transposable element gene,暗示rack1突变体除了自身直接影响miRNA的表达和其靶基因的表达以外,也可能通过影响transposable element gene的表达来更为广泛地影响它们所调节的基因的表达.进一步的分析表明:rack1影响的miR397b和miR5028的靶基因分别是HEMC和HEMB1,它们是参与叶绿体叶绿素合成途径中上游途径的关键步骤中的酶类,暗示RACK1在调控植物的叶绿体的发育和功能中扮演了关键的角色.
猜你喜欢
教学考试(高考生物)(2020年6期)2020-11-23
食品与生物技术学报(2020年8期)2020-01-06
科学24小时(2019年5期)2019-06-11
发明与创新(2019年9期)2019-03-26
知识经济·中国直销(2018年8期)2018-08-23
数学学习与研究(2017年3期)2017-03-09
安徽医科大学学报(2016年12期)2017-01-15
山东农业工程学院学报(2016年6期)2016-12-01
中国老区建设(2016年1期)2016-02-28
天津医科大学学报(2015年2期)2015-12-22
