
Electrochemical Study on Hydrogen Evolution and CO2Reduction on Pt Electrode in Acid Solutions with Different pH
2018-11-09 06:53:20JingYangJieWeiWeiChenYanxiaChen
Jing YangJie WeiWei ChenYan-xia Chen
Hefei National Laboratory for Physical Science at the Microscale and Department of Chemical Physics,University of Science and Technology of China,Hefei 230026,China
Key words:Hydrogen evolution reaction,CO2Reduction,Pt(111),Acidic solution,pH effect,Infrared spectroscopy
I.INTRODUCTION
Electrochemical reduction of CO2(CO2RR)is considered to be a promising approach toward production of value-added chemicals or fuels powered by intermittent renewable energy sources[1,2].In this field,extensive studies have been carried out[3]. Earlier studies on CO2RR on precious metals like Pt and Pd in NaHCO3solution revealed that the major cathodic current(>99%)is from hydrogen evolution reaction(HER)[4].In solution with pH=1,besides the adsorbed CO[5],small amount of formic acid,formaldehyde,and methanol are detected during CO2reduction on Pt[6].However,their current efficiency has not been quantitatively analyzed yet.
In 1990,Bocarslyet al.reported that in pyridine(Pyr)-containing acidic solution with pH close to the pKaof Pyr(Pyr+H+=PyrH+,pKa=5.3),the Faradaic yield for CH3OH generation from CO2reduction on Pt can be up to 30%with HER as the main competitive reaction[7].Since then,extensive studies on Pyr catalyzed CO2RR have been carried out,several mechanisms have also been proposed[2,8].However,a few recent studies of CO2RR on Pt electrodes questioned the catalytic activity of Pyr as well as the reproducibility of earlier results[9–11].Since HER is the major competitive reaction during CO2reduction on Pt with or without molecular electrocatalysts such as Pyr,understanding the electrochemical behavior,the effect of the existence of CO2and its reaction intermediates on HER kinetics will be of great help:(i)to unravel the role of Pyr on CO2RR,(ii)to reveal the viability of using Pt for CO2reduction,(iii)to figure out the optimum conditions for CO2RR electrolysers.We have systematically studied CO2RR on Pt in acid solution with different pH,with or without Pyr and how it competes with HER.In this contribution,we report part of these results of HER on Pt(111)in CO2saturated solution with pH close to the pKaof Pyr.
II.EXPERIMENTS
The electrolyte solutions with 0.1 mol/L NaClO4+xmol/L HClO4(x=10−1,10−2,10−3,3×10−3,10−4,10−5,and 5×10−6mol/L)were prepared using NaClO4(99.99%,Suprapure,SigmaAldrich)andHClO4(70%,Suprapure,Sigma Aldrich)and ultra-pure water(18.2 MΩ·cm,from Mili Q water system).The pH values of the solutions were determined using a pH meter.Before each experiment,all solutions were purged with N2(99.999%,the Linde Group,China)for 20 min.CO2saturated solution was ensured by further purging the solution with CO2(99.99%,the Linde Group,China)for 15 min continuously during the measurements.
Pt(111)and thin Pt film deposited on the flat reflecting face of a hemi-cylindrical Si prism were used as the working electrodes.The preparation,pretreatment,and characterization of Pt(111)electrode were described in detail in Ref.[12].The base CV for the Pt(111)electrode in 0.1 mol/L HClO4is given in FIG.1(a).It displays the well-de fined features reported previously,con firming that the homemade Pt(111)is well-ordered and the cell system used in our study is clean.The Pt thin film electrode with a thickness of ca.50 nm was deposited by electrolessplating,following a procedure described elsewhere[13].The active surface area of the film electrode was ca.3.7 cm2estimated from the charge for the oxidation of a saturated Hadlayer formed in the potential region from 0.4 V to 0.05 V.
A conventional two-compartment electrochemical cell was used for the electrochemical experiment,while the flow cell used for the infrared spectroscopic measurement under attenuated total re flection con figuration is described in detail in Ref.[14].A Pt foil(99.99%)and a reversible hydrogen electrode(RHE)were used as counter and reference electrodes respectively.The measurements with single crystalline electrodes were done under hanging meniscus rotating disk electrode con figuration.The electrode rotation speed was controlled by a modulated rotator(Pine Instruments Company).The electrode potentials were controlled by a potentiostat(CHI700C,Shanghai ChenHua).All potentials are quoted against the RHE.When recording theI-Ecurves,Ohmic compensation is done automatically by the potentiostat.All experiments were carried out at room temperature(ca.25◦C).
III.RESULTS AND DISCUSSION
A.Cyclic voltammetric study on HER on Pt(111)in CO2 saturated electrolyte
FIG.1(b)displays two representative CVs of Pt(111)in 0.1 mol/L NaClO4+5.0×10−6mol/L HClO4with and without saturated CO2(pH=5.3)under stationary condition.For the CV without CO2,the small cathodic peak at ca.0.6 V in the negative-going potential scan is the reductive desorption of OHadthrough
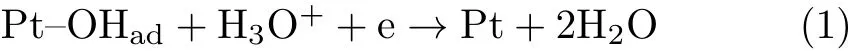
which is followed by the reductive desorption of OHadthrough
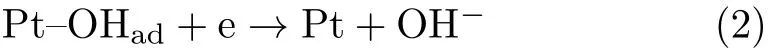
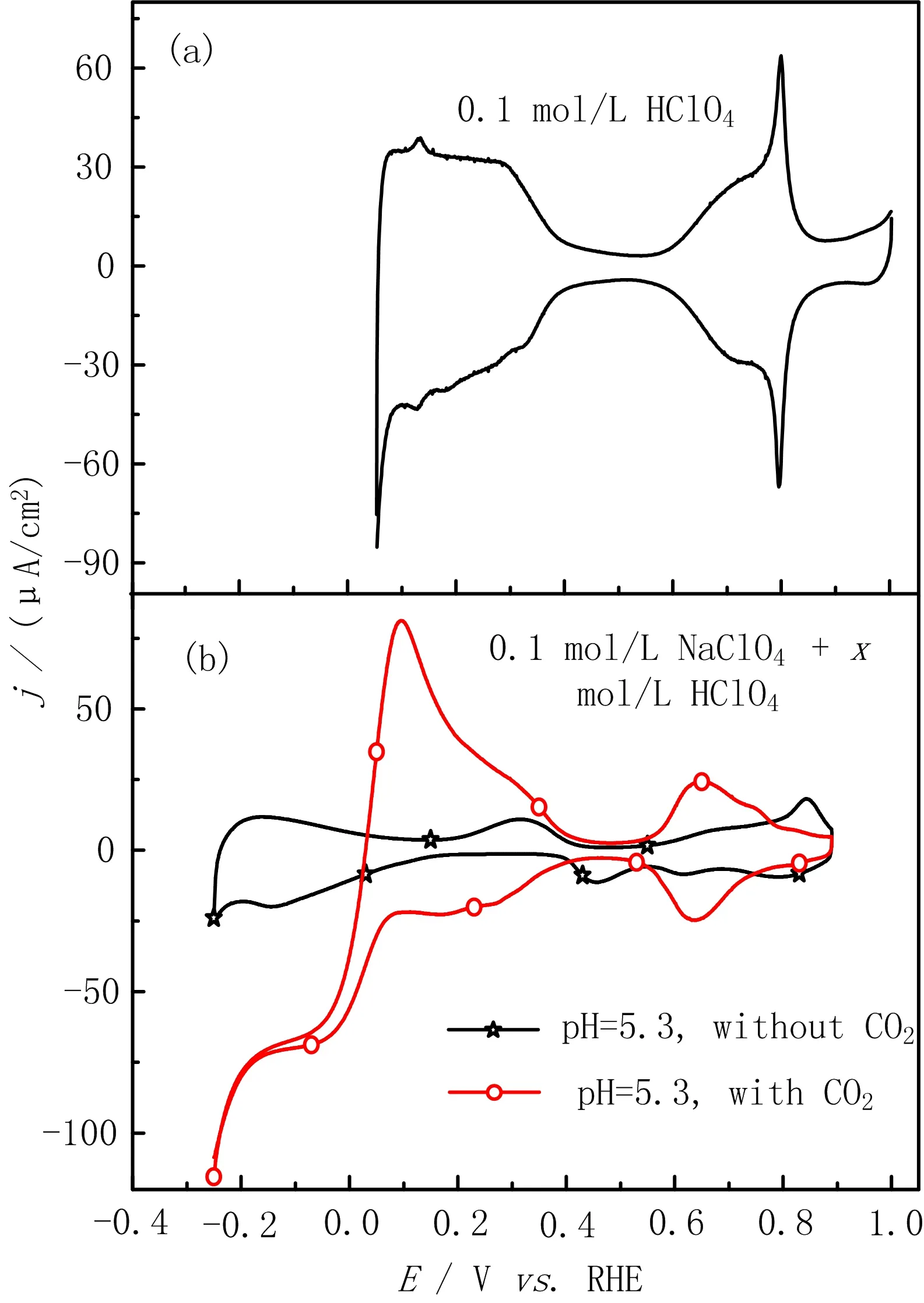
FIG.1 Cyclic voltammogram of Pt(111)in(a)0.1 mol/L HClO4and(b)0.1 mol/L NaClO4+5.0×10−6mol/L HClO4 with(circle)and without(star)CO2,scan rate:50 mV/s,electrode rotation speed:0 r/min.
when the consumption of proton near the electrode surface is much faster than its supply by diffusion from the bulk solution.As a result,the pH near the electrode surface(denoted as pHshereafter)becomes higher than 7,which explains why reaction(2)occurs at more negative potentials than that for reaction(1)[15].As a result of high pHs,the onset potential for under potential deposition(UPD)of H occurs only atE<0.2 V.When the scan direction is reversed at−0.2 V,oxidation of UPD-H occurs immediately.After part of the Hadatoms are oxidized,the pHsdecreases,and the rest of UPD-H is oxidized at higher potentials with a small peak at ca.0.3 V as typical for those in acidic environments.Reaction induced change of the interfacial pH as well as its impact on the related electrode reaction in solution with low H+or OH−concentration and without buffer has been discussed thoroughly by our group previously[15–17].
From the CV recorded in CO2saturated solution,we found that there is a pair of symmetric anodic and cathodic peaks in the potential region from 0.5 V to 0.8 V,which are from the adsorption and desorption of carbonate through
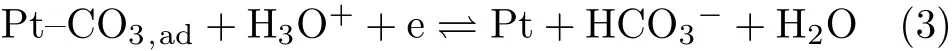
as well con firmed by using infrared spectroscopy[18].The good symmetry of the anodic and cathodic current wave for carbonate adsorption and desorption suggests that the kinetics for its adsorption and desorption is fast.By comparing the CV recorded in CO2saturated solution with the CV given in FIG.1(a),we see that the onset potential for bicarbonate adsorption is ca.50 mV more negative than that for OHadadsorption,whose adsorption will inhibit the OHadadsorption,as similar to the case of acetate adsorption[19].In contract to the case in CO2free solution,in CO2saturated solution,it is found that the current waves for H-UPD and for the oxidation of UPD-H are symmetric,which occur in the potential region as that for the case with pH=1(on the RHE scale)(FIG.1(a)).This indicates that in CO2saturated solution,the pHsnear Pt surface will not change significantly upon the UPD of H due to the buffer capability of CO2even when the pH of the bulk solution is 5.3.WhenEis more negative than 0.1 V,HER through
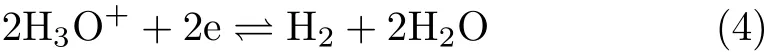
occurs,it increases sharply with the negative shift in potential and reaches a plateau whenEis negative than−0.05 V due to the small H+concentration in the bulk solution with pH=5.3.WhenEis more negative than−0.1 V,the cathodic current increases again,which mainly comes from

Instead of CO2reduction(for further evidence,see text below)[20,21].After scanning to−0.25 V and then reversing the potential scan,anodic current appears atE>0.05 V,with an amplitude much higher than that for UPD-H during the cathodic scan in the same potential region.The extra current comes from H2oxidation,which is formed during previous negative-going scan down to−0.25 V.
In order to further con firm this,CVs with the same scan rate but under different electrode rotation speed or with different scan rate under stationary conditions are recorded,which are shown in FIG.2 and FIG.3.From FIG.2,we see that with the increase of the electrode rotation speed,the current wave in the potential region from 0.05 V to 0.4 V decreases,only H-UPD current is observed when rotation speed is higher than 1600 r/min.Since the oxidation currents for CO2reduction products,such as HCOOH,CO,and CH3OH are very small in the potential region withE<0.4 V[14,22],the anodic current observed in the potential region from 0.05 V to 0.4 V must come from the oxidation of H2formed at negative potential which has not been diffused away from the surface.This is further con firmed by the data given in FIG.3,where we found that with the increase of potential scan rate the pseudo capacitance for bicarbonate adsorption does not change,while that in the H-UPD region decreases.At lower potential scan rate,more time is spent at HER potentials.As a result,more H2are produced(FIG.3(b)),hence more H2are available near the electrode surface to be oxidized in the H-UPD potential region.
Furthermore,we found that the diffusion limiting current for HER through reaction(4)is much larger than what is predicted by the Levich equation,Eq.(6)(inset in FIG.2):
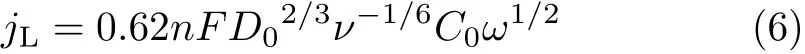

FIG.2 Cyclic voltammogram of Pt(111)in 0.1 mol/L NaClO4+5.0×10−6mol/L HClO4with CO2under different electrode rotation speed,scan rate:50 mV/s.The inset shows the the measured(square)and estimated(circle)diffusion limiting current for HER at−0.1 V with H+as reactant.
wherenis the number of electrons,Fis the Faraday constant,D0is the diffusion coefficient,νis the dynamic viscosity,C0is the bulk concentration,andωis the rotation speed.
The higher HER current is explained by the buffer capability of CO2when the rate of the consumption of proton can be fast compensated by the dissociation of H2CO3.AtE<−0.2 V,the HER current does not show obvious change with electrode rotation speed,which can be explained by the fact that water is the precursor for HER,the reaction is not affected by the mass transport since water concentration in the solution is ca.55 mol/L. The pH of bulk solution is 5.3,only ca.3 pH unit smaller than 8 for a basic solution,which explains why the plateau for HER through reaction(4)only covers a potential region of ca.0.15 V(i.e.,3×0.059 V),agreeing well with the shift of the equilibrium potential for HER according to the Nernst equation.With the increase of potential scan rate,the slight positive shift in HOR current peak or negative shift in the HER current wave(FIG.3)is not due to the uncompensated Ohmic resistance,since the overall current is small and Ohmic compensation is automatically applied during the data recording.Instead,we think it originates from low proton concentration which renders the local pHschange being more pronounced,i.e.,the abrupt change of local pHsinduced by fast consumption or production of H+cannot be compensated by the diffusion.This is further con firmed by the comparison of the CVs recorded in solution with different pH(FIG.4).From FIG.4 we see that with the decrease of solution pH,the current for both HER and HOR at ca.0 V increases,and the anodic and cathodic current peaks become more symmetric.Note that the shift of the current wave for carbonate adsorption is due to the pH induced change of concentration of bicarbonate precursors.The decay of HER current atE<−0.05 V is due to the mass transport limit of H+.All the data discussed above indicate that the characteristic current features atE<0.4 V either as a function of potential scan rate,electrode rotation speed or solution pH agree well with the typical behavior of pH effect on HER and HOR,while the current features for CO2reduction are not obvious at all.
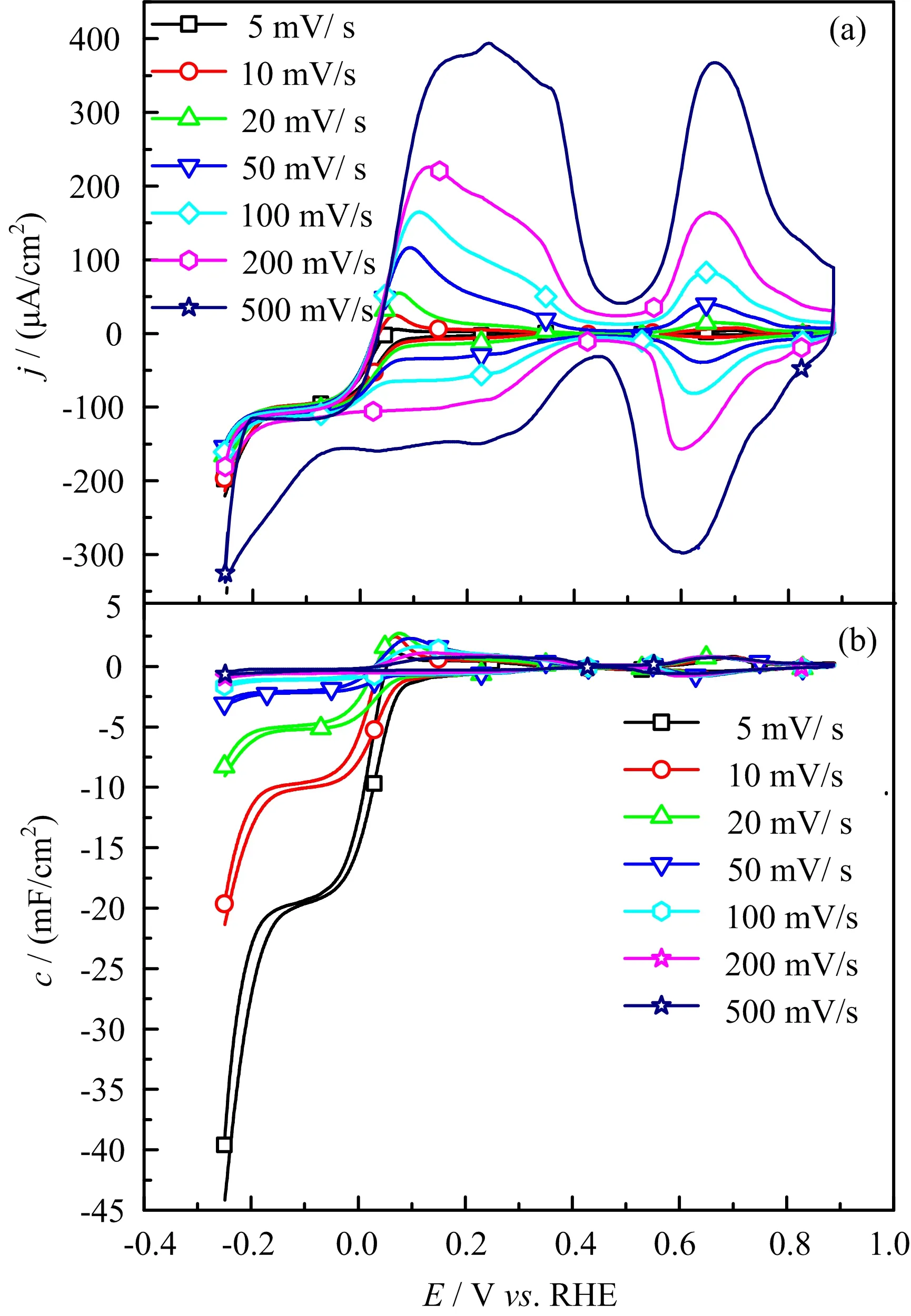
FIG.3(a)Cyclic voltammogram and(b)capacitance of Pt(111)in 0.1 mol/L NaClO4+5.0×10−6mol/L HClO4with CO2under different potential scan rates,electrode rotation speed:0 r/min.
In solutions with low pH(≤1),it is well con firmed by infrared spectroscopy that COadwill be formed at potentials where HER or UPD-H occurs[5,23–26].In order to check whether COador other intermediates may be formed under present condition with high local pH,which may affect the HER or HOR kinetics,a preliminary study was carried out by holding the potential in the HER potential region for 2 min,then examining the CV with and without such holding treatment(FIG.5).We found that the CVs recorded with and without such holding are nearly the same,indicating that both the HER and HOR activities are not affected by any possible intermediates formed during CO2reduction.Slightly higher current at ca.0.7 V which is superimposed on the current wave for carbonate adsorption is probably due to the oxidation of COadon Pt(111).This will be further discussed in Section III.B.The contribution of all simultaneous reactions in our system,such as HER,HOR,and CO2RR,is unclear when only considering the current data.However,such question can be solved by combination of otherin situelectrochemical techniques like differential electrochemical mass spectrometry(DEMS).
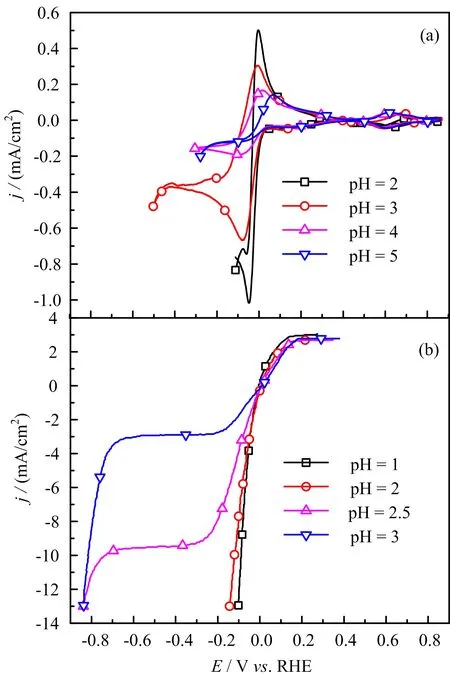
FIG.4 Cyclic voltammogram of Pt(111)in(a)CO2and(b)H2saturated 0.1 mol/L NaClO4+x mol/L HClO4(x=10−1,10−2,10−3,3×10−3,10−4,and 10−5mol/L)of different pH as indicated in the figure,scan rate:50 mV/s,electrode rotation speed for(a)0 r/min and(b)1600 r/min,respectively.
The results discussed in this section can be summarized as follows:(i)in solutions with pH>2,the interfacial pH increases abruptly during HER,which cannot be buffered by CO2;(ii)as a result,HER with H+as reactant occurs at lower overpotentials;while at higher overpotentials,HER with H2O as reactant occurs;(iii)carbonate adsorption is observed in the potential region from 0.55 V to 0.8 V;(iv)the kinetics of both HER and HOR are not affected by the adsorbed intermediates which is formed by CO2reduction;(v)the good symmetry of the anodic and cathodic current waves for carbonate adsorption and desorption suggests that the kinetics for its adsorption and desorption is fast.

FIG.5 Cyclic voltammogram of Pt(111)in CO2saturated 0.1 mol/L NaClO4+x mol/L HClO4(x=10−2,10−3,and 10−4mol/L)of different pH(a)pH=2,(b)pH=3,(c)pH=4,with(star)and without(circle)holding at−0.1 V for 2 min,scan rate:50 mV/s,electrode rotation speed:0 r/min.
B.Infrared spectroscopic study on the interface Pt/CO2 saturated electrolyte and its impact on HER
In order to get more insights into whether CO2reduction also occurs during HER and how the adsorbed intermediates formed from CO2RR affect the HER kinetics,we have carried out electrochemicalin-situinfrared spectroscopic measurements of the Pt interface under attenuated total re flection con figuration(ATRFTIRS).FIG.6 displays the cyclic voltammogram of Pt film in N2or CO2saturated 0.1 mol/L NaClO4+5.0×10−6mol/L HClO4.Note that in order to get IR spectra with good quality,the potential scan rate is limited to 10 mV/s.As similar to the case for Pt(111)in N2saturated solution,due to the low proton concentration as well as lacking of buffer capability,the current wave for H-UPD and its oxidative removal is displaced by ca.0.4 V.In contrast,in CO2saturated solution,due to the buffer capability of CO2,the potential for H-UPD and its oxidation removal is only shifted toward negative values by ca.0.2 V.The anodic current wave in CO2saturated solution is much broader than that in N2saturated solution,which is probably due to the superimposition of H2oxidation.Because of the slow scan rate as well as stationary condition without stirring,the contribution of the current wave from HER and HOR is very obvious.As a result,the current wave for carbonate adsorption/desorption in the potential region from 0.5 V to 0.8 V becomes less obvious.

FIG.6 Cyclic voltammogram of thin Pt film electrode in 0.1 mol/L NaClO4+5.0×10−6mol/L HClO4with(circle)and without(star)CO2,scan rate:10 mV/s.
FIG.7(a)displays the IR spectra of Pt interface at some selected potentials in N2saturated solution recorded simultaneously with the CV given in FIG.7.From FIG.7(a)we see that there is barely no spectral features atE>0 V.WhenEis below 0 V,only the bending(1645 cm−1)and stretching mode(3000−3600 cm−1)[27]of water are observed,whose band intensity increases with decreasing the electrode potential. Such water species are assigned to water molecules which form hydrogen bonds with UPD-H atoms adsorbed on Pt surface,its band intensity displays roughly a linear relationship with the coverage of UPD-H atoms,similar to previous observations in solution with pH=1[28,29].
In the CO2saturated solution,again,the positivepointing OH stretching appears in the H-UPD potential region,while that for the water bending is not obvious.This is probably due to the appearance of the negative-pointing carbonate band at ca.1545 cm−1[30,31],which appears at ca.0.7 V,whose band intensity increases with further negative shift in potential and reaches the maximum at ca.0.4 V,in good agreement with the indications given by CV(FIG.1 and FIG.6).This con firms that the carbonate is desorbed atE<0.5 V.Besides the water band,two positive bands at 1770 and 1989 cm−1appear whenEis below 0.2 V.These bands are assigned to adsorbed CO in multiply-bonded(COM)and linearly-bonded(COL)con figuration,respectively[27].In the reverse scan from−0.4 V to 0 V,both the band intensity and the peak frequencies of the CO band increase further with the positive shift in electrode potential.At higher poten-tials,the band intensity for COLincreases while that for COMdecreases,indicating there is a transfer of COMto COL.AtEabove 0.4 V the intensity of both bands decreases again and finally disappears whenEis above 0.8 V,which indicates that COadis oxidized at higher potentials.The IR data reveal that in solution with pH=5,COadcan be formed from CO2reduction in the potential region where H-UPD or HER occurs,similar to the case with pH=1[5].Besides the COad,no other adsorbed intermediates are detected by ATR-FTIRS in the potential region where H-UPD or HER occurs.
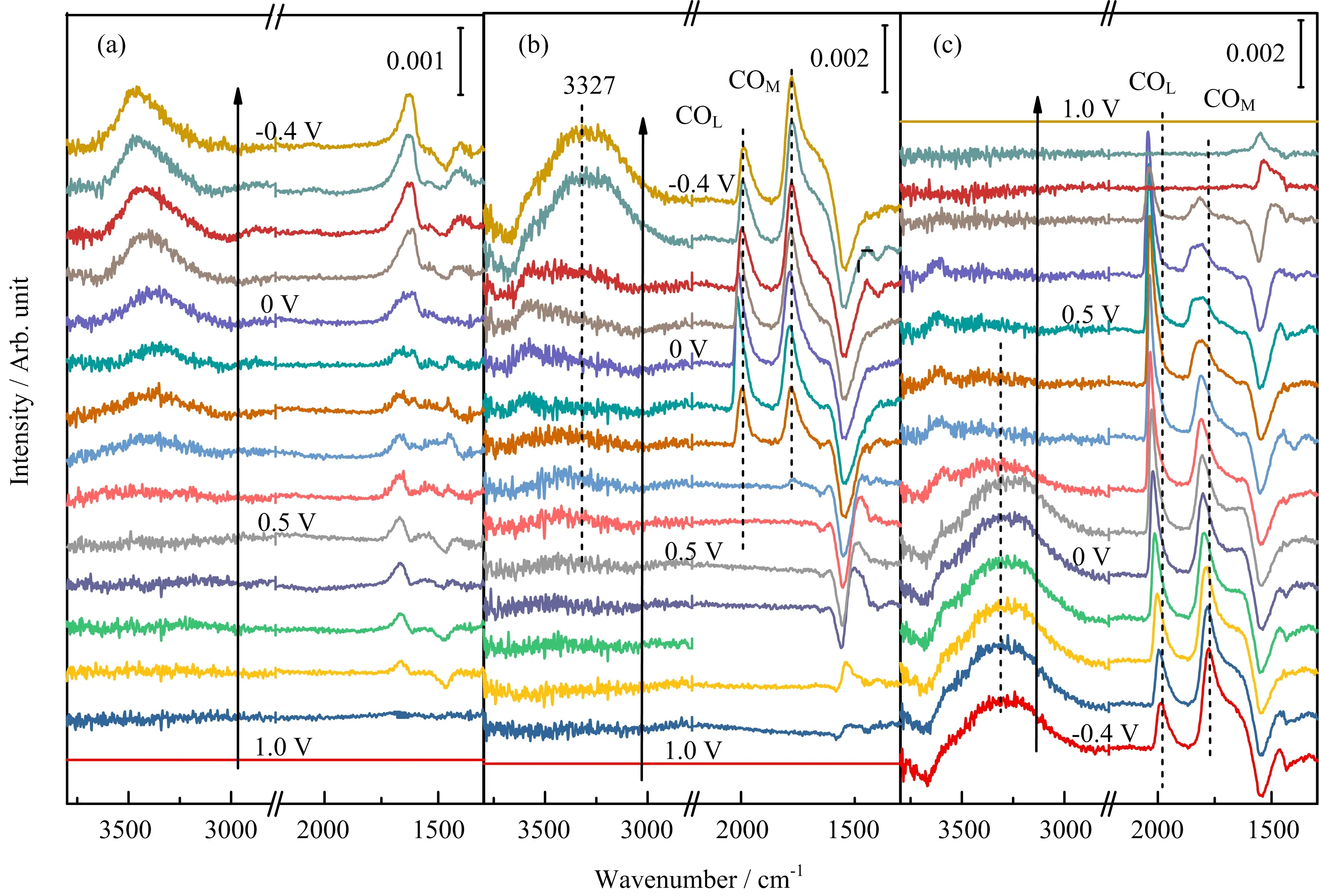
FIG.7 Selected IR spectra at Pt film electrode in N2(a)and CO2(b,c)saturated 0.1 mol/L NaClO4+5.0×10−6mol/L HClO4recorded during cyclic voltammetric potential scan,scan rate:10 mV/s

FIG.8 Current transients for HER and CO2RR on Pt film electrode after stepping the potential from 0.9 V to 0.3 V(square),0 V(circle),and−0.3 V(triangle).
In order to follow the kinetics for COadformation on Pt film electrode during H-UPD or HER,IR spectra were recorded with current transients upon stepping the electrode potential from 0.9 V to 0.3 V,0 V,or−0.3 V,and after holding at that potential for 30 s the potential is step back to 0.9 V again.The current transients are displayed in FIG.8 and the time-dependent IR spectra are shown in FIG.9.From FIG.8 we see that right after stepping the potential to 0.3 V or 0 V,only current for double layer charging was observed,and it decreases fast to zero.When stepping to−0.3 V,there is a large cathodic current right after the potential step,and it decreases fast with reaction time and reaches steady state at ca.20 s after the potential step.From the simultaneously recorded IR spectra,we found that the rate for COadbuild up is the slowest at 0.3 V and the fastest at 0 V.So far,it is not clear whether the amount of adsorbed H plays a role for such a difference,or it is just due to the change of thermodynamic driving force for CO2RR.For the case of stepping to−0.3 V,more COMis formed than that for COL,the IR band intensity of the COLand COMbands first increases with reaction time up to 20 s,then it does not increase anymore,although the overall COadsurface coverage is below 0.15 ML[5].This coincides with the fact that the current decreases with reaction time and it reaches steady state also at ca.20 s after potential step.Further studies are necessary to figure out whether this is because CO2RR to COadneeds special active sites which are fully occupied by COador HER.The limited COadformation under HER is probably due to dynamic turnover of Hadon the surface which limits the available sites and residence time for the reduction intermediates(Had)necessary for CO2adsorption and reduction.

FIG.9 Selected IR spectra at Pt film electrode in CO2saturated 0.1 mol/L NaClO4+5.0×10−6mol/L HClO4recorded during stepping the potential from 0.9 V to 0.3 V(a),0 V(b),and−0.3 V(c).
An interesting phenomenon we would like to point out here is that although the IR band intensities of the COadbands do not show obvious increase further with reaction time at ca.20 s after the potential is stepped to−0.3 V,the peak frequencies of the COadbands still display a continuous increase with reaction time(FIG.10).Similar increase of CO stretching frequency in the H-UPD potential region was observed before,but it was accompanied by an increase of the band intensity.The latter is explained by the adsorbed H-induced CO migration and COadislands formation.The enhanced dipole-dipole coupling between adsorbed CO at the neighboring sites is suggested to be the origin for the increase of both COadband intensity and peak frequency[32].However,in the HER region,as observed in present study only the increase of peak frequency is obvious,while that of CO band intensity is not.Our preliminary explanation for this is that under HER condition,CO also diffuses along Pt surface until it finds the proper sites.The electronic effect of adsorbed H in the neighboring sites may lead to less electrons transfer from Pt to the anti-bonding orbital of CO bond.As a result,the CO stretching frequency increases.A schematic illustration of surface adsorption of CO is given in FIG.11 for better understanding.Further studies with DFT calculations are underway to verify this.
IV.CONCLUSION
The competition of hydrogen evolution reaction and CO2reduction on Pt electrode is investigated by cyclic voltammetry and infrared spectroscopy. We found that in solution with pH>2,the interfacial pH increases abruptly during HER.As a result,HER with H+as reactant occurs at lower overpotentials,while at higher overpotentials,HER with H2O as reactant occurs.COadcan be formed by CO2reduction on Pt at potentials where UPD-H or HER occurs.The rate for COadformation increases with the coverage of UPD-H and reaches its maximum at the onset potential for HER.The decrease of COadformation under HER is attributed to the limited sites available and limited residence time for the reduction intermediates(Had)necessary for CO2adsorption and reduction.Furthermore,we found that under HER condition,the peak frequency for COadincreases continuously with reaction time,while its band intensity does not.The diffusion of COadon Pt surface introduced by the dynamic turnover of Hadis suggested to be the origin for such change.
V.ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China(No.21473175 and No.21273215)and the Ministry of Science and Technology of China(No.2015CB932301).
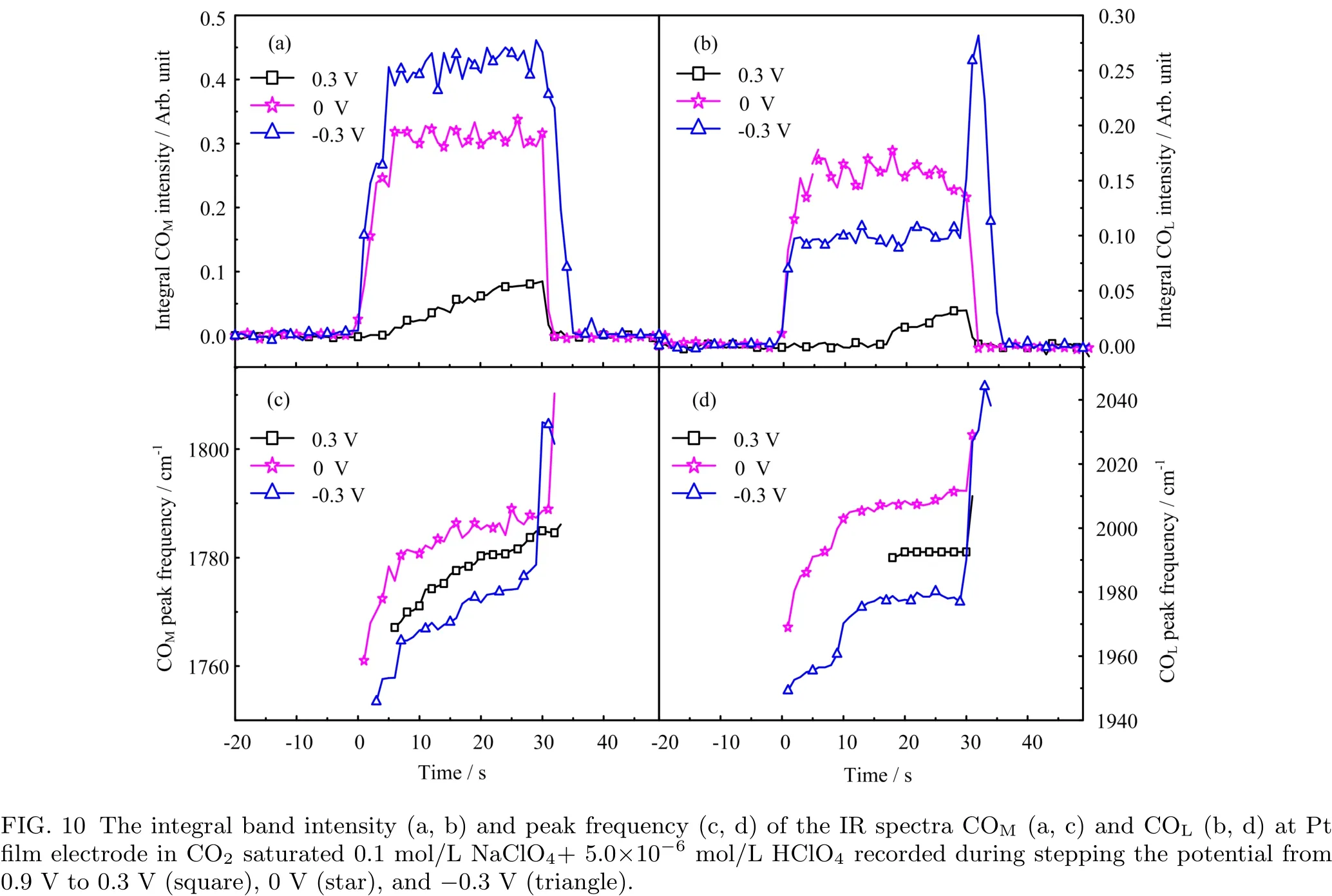
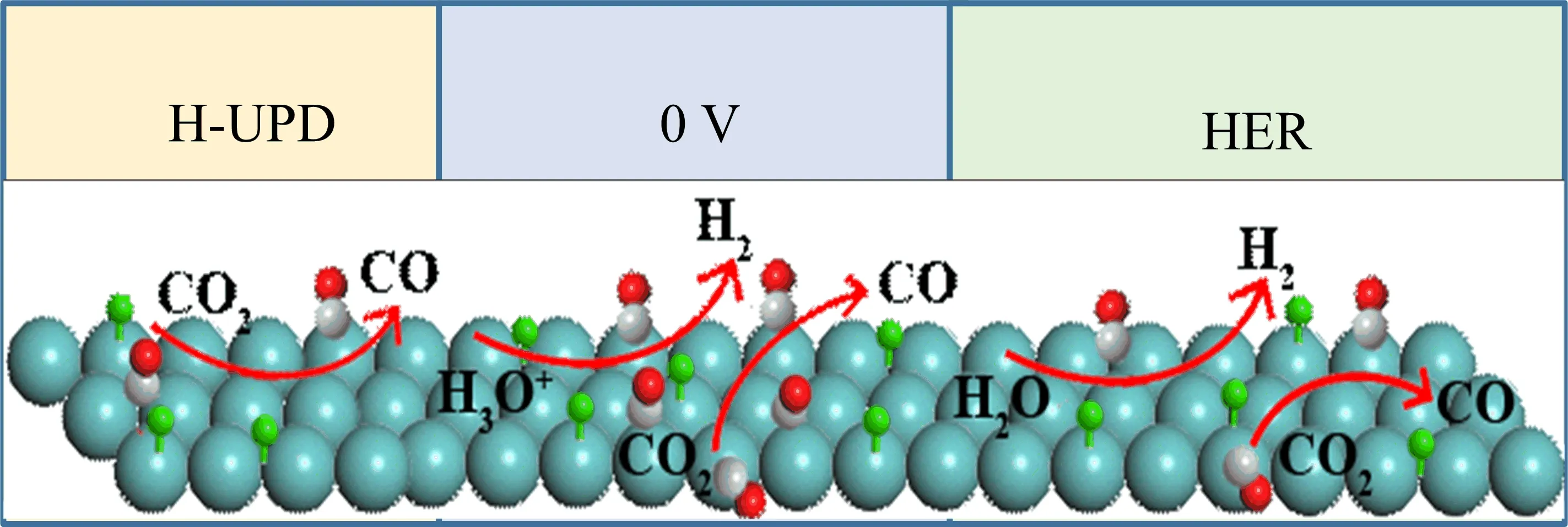
FIG.11 Schematic illustration of reaction at Pt electeode in CO2saturated solution in different potential regions.
[1]H.R.M.Jhong,S.Ma,and P.J.Kenis,Curr.Opin.Chem.Eng.2,191(2013).
[2]R.Francke,B.Schille,and M.Roemelt,Chem.Rev.(2018).
[3]Y.Hori,Modern Aspects of Electrochemistry No.42,C.G.Vayenas,R.E.White,and M.E.Gamboa-Aldeco,Eds.,New York,NY:Springer,89(2008).
[4]Y.Hori,H.Wakebe,T.Tsukamoto,and O.Koga,Electrochim.Acta 39,1833(1994).
[5]T.Smolinka,M.Heinen,Y.X.Chen,Z.Jusys,W.Lehnert,and R.J.Behm,Electrochim.Acta 50,5189(2005).
[6]A.A.Ensa fi,H.A.Alinaja fi,and B.Rezaei,J.Electroanal.Chem.783,82(2016).
[7]G.Seshadri,C.Lin,and A.B.Bocarsly,J.Electroanal.Chem.372,145(1994).
[8]E.B.Cole,P.S.Lakkaraju,D.M.Rampulla,A.J.Morris,E.Abelev,and A.B.Bocarsly,J.Am.Chem.Soc.132,11539(2010).
[9]C.Costentin,J.C.Canales,B.Haddou,and J.M.Saveant,J.Am.Chem.Soc.135,17671(2013).
[10]C.X.Kronawitter,Z.Chen,P.Zhao,X.Yang,and B.E.Koel,Catal.Sci.Technol.7,831(2017).
[11]H.Dridi,C.Comminges,C.Morais,J.C.Meledje,K.B.Kokoh,C.Costentin,and J.M.Saveant,J.Am.Chem.Soc.139,13922(2017).
[12]J.Xu,D.Yuan,F.Yang,D.Mei,Z.Zhang,and Y.X.Chen,Phys.Chem.Chem.Phys.15,4367(2013).
[13]N.Hoshi,E.Sato,and Y.Hori,J.Electroanal.Chem.540,105(2003).
[14]W.Chen,J.Cai,J.Yang,M.M.Sartin,and Y.X.Chen,J.Electroanal.Chem.800,89(2017).
[15]L.W.Liao,S.X.Liu,Q.A.Tao,B.Geng,P.Zhang,C.M.Wang,Y.X.Chen,and S.Ye,J.Electroanal.Chem.650,233(2011).
[16]M.F.Li,L.W.Liao,D.F.Yuan,D.Mei,and Y.X.Chen,Electrochim.Acta 110,780(2013).
[17]D.Mei,Z.D.He,Y.L.Zheng,D.C.Jiang,and Y.X.Chen,Phys.Chem.Chem.Phys.16,13762(2014).
[18]R.Martinez-Hincapie,A.Berna,A.Rodes,V.Climent,and J.M.Feliu,J.Phys.Chem.C 120,16191(2016).
[19]X.Q.Zuo,W.Chen,A.Yu,M.Le Xu,J.Cai,and Y.X.Chen,Electrochem.Commun.89,6(2018).
[20]V.Grozovski,S.Vesztergom,G.G.Láng,and P.Broekmann,J.Electro.Chem.Soc.164,3171(2017).
[21]D.Strmcnik,P.P.Lopes,B.Genorio,V.R.Stamenkovic,and N.M.Markovic,Nano Energy 29,29(2016).
[22]Y.Wei,X.Q.Zuo,Z.Da He,W.Chen,C.H.Lin,J.Cai,M.Sartin,and Y.X.Chen,Electrochem.Commun.81,1(2017).
[23]S.G.Sun and Z.Y.Zhou,Phys.Chem.Chem.Phys.3,3277(2001).
[24]A.Rodes,E.Pastor,and T.Iwasita,J.Electroanal.Chem.369,183(1994).
[25]A.Rodes,E.Pastor,and T.Iwasita,J.Electroanal.Chem.377,215(1994).
[26]A.Rodes,E.Pastor,and T.Iwasita,J.Electroanal.Chem.373,167(1994).
[27]S.X.Liu,L.W.Liao,Q.Tao,Y.X.Chen,and S.Ye,Phys.Chem.Chem.Phys.13,9725(2011).
[28]K.Ataka,T.Yotsuyanagi,and M.Osawa,J.Phys.Chem.100,10664(1996).
[29]M.Osawa,M.Tsushima,H.Mogami,G.Samjeske,and A.Yamakata,J.Phys.Chem.C 112,4248(2008).
[30]A.Berna,A.Rodes,J.M.Feliu,F.Illas,A.Gil,A.Clotet,and J.M.Ricart,J.Phys.Chem.B 108,17928(2004).
[31]T.Iwasita,A.Rodes,and E.Pastor,J.Electroanal.Chem.383,181(1995).
[32]Y.X.Chen,M.Heinen,Z.Jusys,and R.J.Behm,J.Phys.Chem.C 111,435(2007).
 CHINESE JOURNAL OF CHEMICAL PHYSICS2018年5期
CHINESE JOURNAL OF CHEMICAL PHYSICS2018年5期
- CHINESE JOURNAL OF CHEMICAL PHYSICS的其它文章
- A Simple,Compact and Rigid Scanning Tunneling Microscope
- Extraction of Lignin from Tobacco Stem using Ionic Liquid
- Gd Doped Hollow Nanoscale Coordination Polymers as Multimodal Imaging Agents and a Potential Drug Delivery Carriers
- 3D Macro-Micro-Mesoporous FeC2O4/Graphene Hydrogel Electrode for High-Performance 2.5 V Aqueous Asymmetric Supercapacitors
- Gamma Ray Radiation Effect on Bi2WO6Photocatalyst
- Ag-Cu Nanoparticles Supported on N-Doped TiO2Nanowire Arrays for Efficient Photocatalytic CO2Reduction