
GC在常见催眠镇静类药物的快速识别中的应用
2018-11-08 02:54:44房永飞
心血管外科杂志(电子版) 2018年1期
房永飞
(上海铁路公安局刑事技术处,上海 200071)
在检测未知催眠镇静类药物时,通常可选用GC、HPLC、GC/MS、GC/FIR、HPLC/MS等多种仪器分析方法来最终确定药物的成分。然而当工作开始时,因检验人员一时不清楚被检药物的种类,往往根据主观判断先选择一种方法进行检测,当发现不适合时再换另一种方法[1]。这样工作盲目性很大,效率低,耗时长,而且不同药物的检验方法差别也比较大,必须另辟蹊径[2]。本文尝试建立一种快速识别此类药物的通用方法,可把未知药物待测定成分初步判定为一种或少数几种标准药物之一,为后续开展优选方法、设定参数及条件等仪器分析工作提供参考依据。
1 实验材料与方法
1.1 仪器 HP5890II型气相色谱仪(配备FID检测器),HP3365化学工作站,GCD-300A氢气发生器,25L空气压缩机。
1.2 标准药物与试剂 巴比妥、戊巴比妥,盐酸哌替啶、盐酸普鲁卡因、强痛定、盐酸吗啡、盐酸氯丙嗪、苯巴比妥钠、硫喷妥钠、眠尔通、安眠酮、扑痫酮、苯妥英钠(大仑丁)、可待因、安定、硝基安定、利眠宁、舒乐安定(艾司唑仑)、氯硝安定、三唑仑、海洛因。甲醇(分析纯),蒸馏水。
1.3 实验条件 OV-1交联甲基硅酮弹性石英毛细管色谱柱(15 mm×0.53 mm×0.52 μm)(八方分析测试新技术研究所);进样温度280oC;FID检测器温度285oC;炉温:150 V(保持0.1 min),程序升温,速率15oC/min,到达225oC(保持3.5 min),再程序升温,速率10oC/min,终止温度255oC(保持10 min);载气为氮气,流量6.5 mL/min;分流进样,分流比13:1;尾吹气30 mL/min;氢气20 mL/min;空气400 mL/min[3]。
1.4 样品前处理及仪器分析 取片剂药物1/3片-1/2片,粉剂药物50 mg左右,粉碎,置于5 mL试管中,加入4 mL甲醇提取,充分混匀,离心,吸取上清液置于5 mL试管中供GC分析;吸取针剂药物溶液2滴,置于5 mL试管中,加入2 mL-2.5 mL甲醇,混匀,供GC分析。取21个药物溶液分别进样,进样量1 μL,记录相同条件下的出峰时间。
2 实验结果与讨论
2.1 按保留时间(出峰顺序)先后列表(表1)
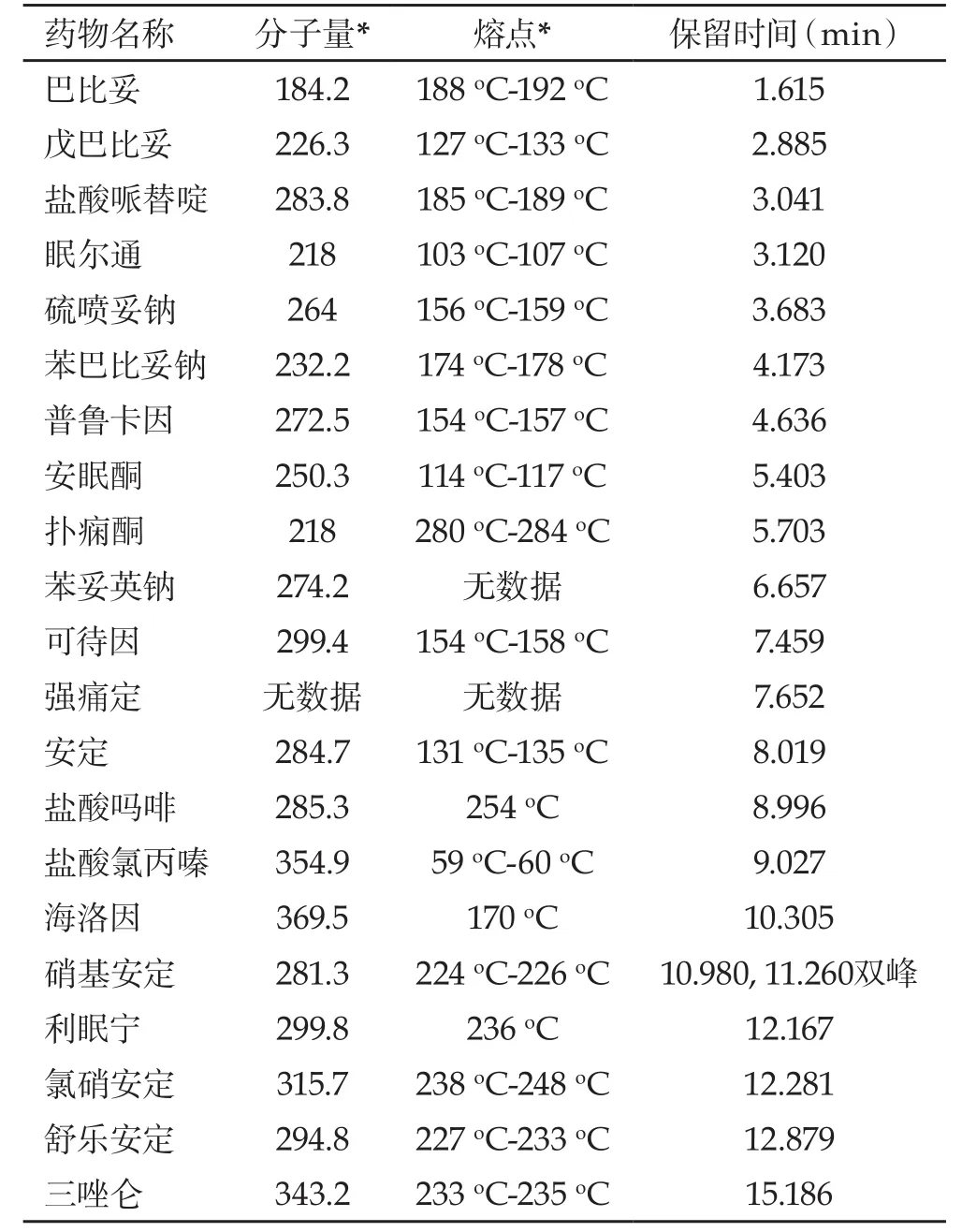
表 1 被检测药物保留时间
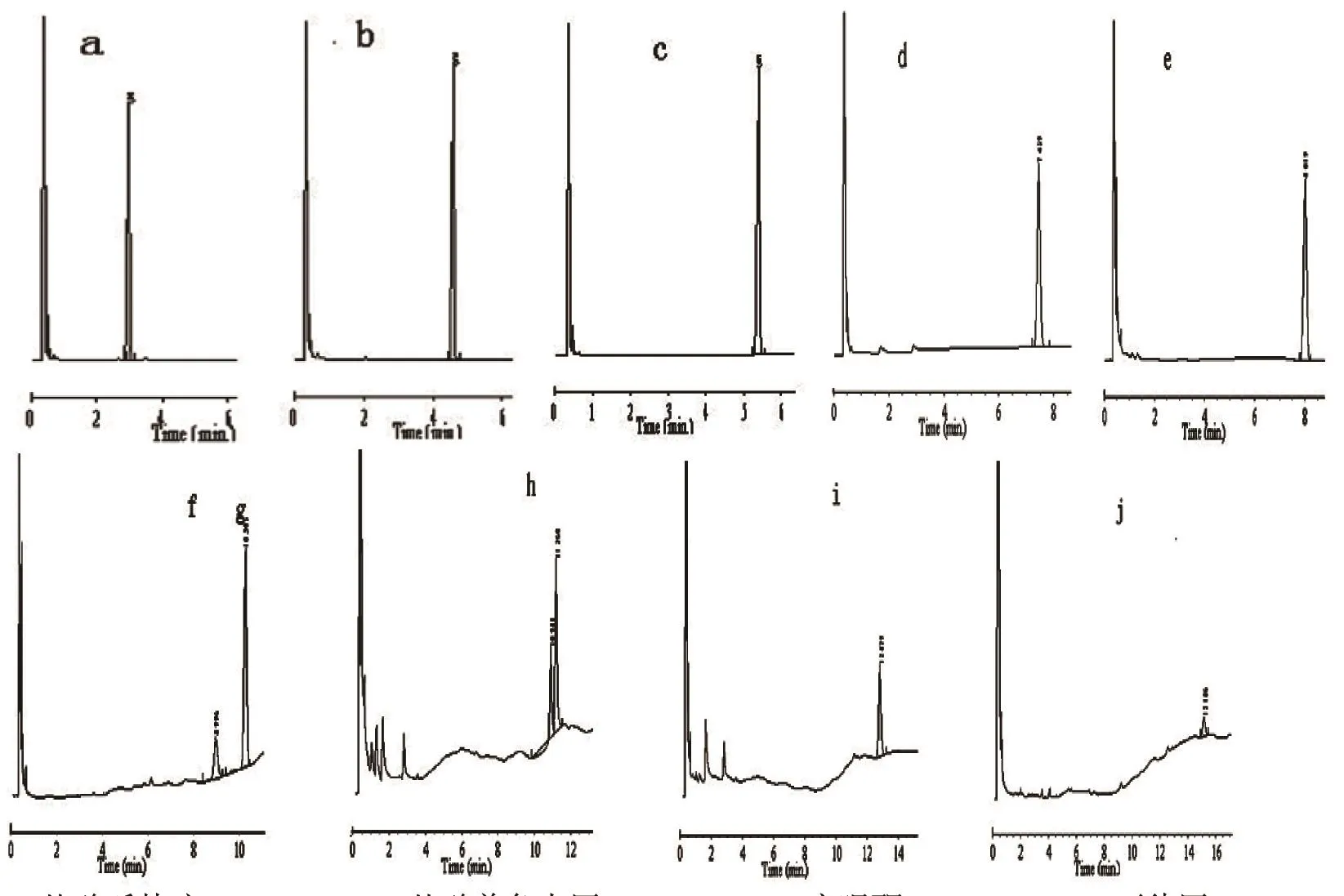
图 1 部分药物色谱图。a.盐酸哌替啶:3.041 min,b.盐酸普鲁卡因:4.636 min,c.安眠酮:5.403 min,d.可待因:7.459 min,e.安定:8.019 min,f.吗啡:8.996 min,g.海洛因:10.305 min,h.硝基安定:10.980/11.260 min,i.舒乐安定:12.879 min,j.三唑仑:15.186 min。
2.2 部分标准药物气相色谱图(图 1)
2.3 数据分析 从表1可以看出,大部分此类药物分子量较大,通常在250以上;熔点较高,部分可达200oC以上。影响保留时间的因素是多方面的,通常在同类化合物中,如果分子量较大、熔点较高,该化合物的保留时间也较长,即出峰较困难。
2.4 实验条件选择 大多数催眠镇静类药物是分子量较大、熔点较高的难挥发性物质,应选择较高的炉温(通常在200oC以上),才能够顺利出峰。实验选择非极性的OV-1大口径毛细管柱,主要是因为固定液的工作温度较高(可达325oC),能满足较大范围内的炉温变化需要;色谱柱口径大也能缩短样品出峰时间。考虑到海洛因出峰时间处于比较中间的位置,通过调节柱头压,把海洛因出峰时间控制在10 min左右。
2.5 分析方法的评论
2.5.1 重现性 实验中标准品的保留时间日间偏差大于日内偏差。同一标准品多次进样后的保留时间,最大日间偏差为0.15 min,最大日内偏差为0.05 min,重现性较好。
2.5.2 定性 本法采用标准品对照法定性,主要侧重对未知样品的快速筛选,以期集中到1个-2个或少数几个标准品的确认上。最终认定可以通过变换实验条件或更换不同极性色谱柱的方法来实现:如把程序升温速率减至5oC/min,就能延长样品保留时间,使两个样品得以分辨;若减小载气流量,也能延长其保留时间;另外,还可以通过改用极性色谱柱、增加对样品的选择性来达到分辨的目的。
2.5.3 提取溶剂的选择 此类药物在甲醇中都有一定的溶解度,而药片和药粉基质部分则基本不被提取,对色谱峰干扰较少,从实验获得的色谱图可得到验证。
2.5.4 检出率 采用本法检测此类药物,主要取决于实验室掌握的标准品的多少。尽可能多地收集此类药物标准品,可以提高送检的检出率。
2.6 低沸点或热不稳定药物的处理 少数催眠镇静类药物沸点则较低、易于挥发,在上述条件下出峰较早(一般在2 min以内),易受溶剂峰或杂质峰干扰;它们的保留时间靠得太近,难以区分。有些药物受热还容易分解。
对此应该降低炉温,例如起始温度选择100oC上下,终止温度设在200oC以内,这样就能在相对较低的温度下进行分析,样品保留时间也会延长。对少数受热易分解的药物,必要时可以将其衍生化,使其生成较高沸点的衍生物,再用本文开头介绍的方法进行分析。
3 结论
本文基于在开始分析未知催眠镇静类药物时遇到的如何优选方法的问题,利用气相色谱技术建立一种快速识别未知药物成分或缩小检测范围的方法,为后续选定最佳方法和开展相关仪器分析工作提供了可靠的参考依据。该方法经实验室盲样测试和实际办案检验,存在灵敏度高、选择性好等优点,且同一保留时间对应两种及以上药物的情况并不多见,具有较高的应用和推广价值。
猜你喜欢
环球时报(2022-05-18)2022-05-18 15:22:01
山西化工(2020年6期)2021-01-10 03:16:40
合成化学(2015年4期)2016-01-17 09:01:02
中国卫生标准管理(2015年25期)2016-01-14 09:29:24
湖南中医药大学学报(2015年1期)2016-01-06 01:06:36
吉林大学学报(医学版)(2015年1期)2015-12-17 07:47:30
中国卫生标准管理(2015年15期)2015-01-26 20:32:38
中国氯碱(2014年10期)2014-02-28 01:04:57
环境科学导刊(2012年4期)2012-01-29 06:09:30
食品工业科技(2011年12期)2011-11-02 08:37:16
