
Transcriptomic comparison to identify rapidly evolving genes in Braya humilis
2018-10-31 11:50YuMingWeiXiaoFeiMaPengShanZhao
YuMing Wei , XiaoFei Ma , PengShan Zhao *
1. Animal Husbandry Pasture and Green Agriculture Institute of Gansu Academy of Agricultural Sciences, Lanzhou,Gansu 730070, China
2. Key Laboratory of Stress Physiology and Ecology in Cold and Arid Regions, Northwest Institute of Eco-Environment and Resources, Chinese Academy of Sciences, Lanzhou, Gansu 730000, China
ABSTRACT The Brassicaceae species Braya humilis shows broad adaptation to different climatic zones and latitudes. However, the molecular adaptation mechanism of B. humilis is poorly understood. In China, B. humilis is mainly distributed on the Qinghai-Tibetan Plateau (QTP) and in the adjacent arid region. Previous transcriptome analysis of B. humilis has revealed that 39 salt and osmotic stress response genes are subjected to purifying selection during its speciation. To further explore the adaptation mechanism of B. humilis to an arid environment, OrthoMCL program was employed in this study and 6,268 pairs of orthologous gene pairs with high confidence were obtained between B. humilis and Arabidopsis thaliana. A comparative evolutionary analysis based on nonsynonymous to synonymous substitution ratio (Ka/Ks) was then conducted. There were 64 pairs exhibiting a Ka/Ks ratio more than 0.5 and among which, three instrumental candidate genes, T2_20487,T2_22576, and T2_13757, were identified with strong selection signatures (Ka/Ks >1). The corresponding A. thaliana orthologs are double-stranded RNA-binding domain protein, MADS-box family protein, and NADH-dehydrogenase subunit 6, which is encoded by mitochondria genome. This report not only demonstrates the adaptation contribution of fast evolving nuclear genes, but also highlights the potential adaptive value of mitochondria gene to the speciation and adaptation of B. humilis toward the extreme environment in an arid region.
Keywords: Braya humilis; positive selection; Ka/Ks; mitochondria gene variation; NADH-dehydrogenase subunit 6
1 Introduction
The Qinghai-Tibetan Plateau (QTP) is the highest and largest plateau in the world and is very sensitive to global climate change (Zheng, 1996). Plants in the QTP and adjacent arid regions have evolved a variety of phenotypes and physiological and biochemical mechanisms for adaptation to different environmental stresses. Unfortunately, the dissection of their molecular adaptation mechanism is not well known, partially due to a lack of genomic information (Zheng,1996; Niuet al., 2013; Zhanget al., 2013b). With the recent advancement of next generation sequencing, it is now possible to combine gene discovery with characterization of transcript expression. In addition, transcriptome analysis has also been performed to study the evolutionary patterns of genes by calculation of nonsynonymous to synonymous substitution (Ka/Ks)ratios between orthologous genes in closely related plant species (Koeniget al., 2013; Niuet al., 2013;Zhanget al., 2013a; Liuet al., 2014; Chenet al.,2015). Genes with a high divergence are inferred to have experienced rapid evolutional selection (e.g., domestication) or responsible for differentiated environment adaptation (Koeniget al., 2013; Zhanget al.,2013a; Liuet al., 2014; Chenet al., 2015). Thus,comparative transcriptomic analysis will deepen our understanding of how natural populations adapt to diverse environment conditions in arid regions.
Braya humilisis a member of the Brassicaceae family and has a broad distribution across the arcticalpine regions of northern North America and also in northern Asia and Central Asia (Al-Shehbaz and O'Kane, 1997). Of these regions, Central Asia has been suggested as the primary diversity center of theBrayagenus (Warwicket al., 2004). Our wild survey found thatB. humilisis mainly distributed on the QTP and in adjacent arid regions in China, and the phenotypes, including leaf morphology, flower color, and flowering time, are different among different regions.Physiological experiments have also shown thatB.humilisplants can withstand a highly osmotic stress treatment (Wanget al., 2016).
Molecular data demonstrate thatB. humilisis a member of lineage three of the Euclidieae tribe, whileArabidopsis thaliana,A. lyrata, andCapsella rubellaare all classified into lineage one, and the extremophyteThellungiella parvulais included in lineage two(Warwicket al., 2007; Couvreuret al., 2010; Franzkeet al., 2011). Previous transcriptome analysis of aB.humilissample from the arid region has revealed that more than 72% transcripts have top hits with proteins of the Brassicaceae species (Zhaoet al., 2017). A set of highly conserved genes have been identified betweenB. humilisand four other sequenced Brassicaceae species (i.e.,A. thaliana,A. lyrata,C.rubella, andT. parvula) using the reciprocal best blasting program, and among which, 39 genes are involved in salt and osmotic stress responses. Genetic and expression analysis against the orthologs in the model plantA. thalianademonstrate that these 39 genes are under purifying selection and functional conservation inB. humilis(Zhaoet al., 2017). These results suggest that some of the same genetic components are likely employed inB. humilisto adapt to an arid environment (Zhaoet al., 2017). To further explore the adaptation mechanism ofB. humilis, a whole transcriptome analysis versusA. thalianawas performed using the OrthoMCL program in this study.The comparative evolutionary analysis based on Ka/Ks ratio was conducted to identify rapidly evolving genes inB. humilis.
2 Materials and methods
2.1 B. humilis transcriptome data
B. humilisseeds were sampled from the Baihushan Mountain (35°56'21"N, 104°08'13"E, altitude:1,805–1,979 m) in Gansu Province, northwestern China. Average annual precipitation of Yuzhong County, where the sample size is located, is 318 mm.Phenotypes of the adult plant are presented in Figure 1.Transcriptome data from Zhaoet al. (2017) was used for the evolutionary analysis in this study. Briefly,fresh tissues ofB. humilisseedlings grown in the laboratory were collected to extract the total RNA.A complementary DNA library was developed and subsequently sequenced using the Illumina HiSeq2000 platform with paired-end reads. Please refer to Zhaoet al. (2017) for detailed information of RNA extraction, library construction, andde novoassembly.

Figure 1 Photographs of habitat and morphology of B. humilis. The habitat of B. humilis at the foot (a), middle (b), and top (c) of Baihushan Mountain, (d–f) Morphology of inflorescence, flower, and silique
2.2 Ka/Ks analysis
The coding and untranslated regions of eachB.humilistranscript were predicted based on the annotation results in Zhaoet al. (2017).A. thalianatranscriptome and proteome were downloaded from the TAIR10 release (www.arabidopsis.org). Using protein sequences ofB. humilisandA. thaliana, othologous genes were clustered by OrthoMCL software (Liet al., 2003) and the 1:1 orthologous gene pairs were screened as following analysis sequences. Orthologous protein sequences were then aligned with MUSCLE (Edgar, 2004) and all alignments were optimized by Gblocks program to remove problematic sites or regions (Castresana, 2000). Finally, the corresponding nucleic acid sequences of orthologous protein pairs were used to estimate Ka/Ks ratios with the Codeml program of the PAML package (Yang,2007).
2.3 Gene ontology (GO) annotation enrichment analysis
TheA. thalianagene in each orthologous pair with a Ka/Ks ratio >0.5 was subjected to GO annotation enrichment analysis using the default settings in Classification SuperViewer (Provartet al., 2003). The enrichment threshold forp-values was less than 0.05 for the normalized class scores.
3 Results
To investigate the evolutionary selection patterns ofB. humilisgenes, the OrthoMCL program was firstly conducted to identify the orthologous pairs betweenB. humilisandA. thaliana. A total of 6,268 orthologous pairs were chosen to assess the Ka/Ks ratios (Figure 2). The results show that 2,360 pairs of orthologs had Ka/Ks ratios <0.1, and 3,844 pairs had Ka/Ks ratios ranging from 0.1 to 0.5. In total, approximately 99% pairs (6,204) of orthologs had Ka/Ks ratios <0.5, suggesting these genes have evolved under strong selection constraints during the speciation period.
There were 64 pairs of orthologs with Ka/Ks ratios >0.5 (Table 1). Among which, two NADH-dehydrogenase subunit genes (NADs) and two cytochrome c biogenesis genes (CCBs) were included.Some genes that are putatively involved in protein modification and turnover were also included, such as cullin family proteins, F-box proteins, andsmall ubiquitin-related modifier 5(SUMO5). Most significantly, three rapid evolving genes with Ka/Ks ratios >1 were identified and these sequences encoded proteins were NADH-dehydrogenase subunit 6 (NAD6;T2_13757 vs. ATMG00270.1), Double-stranded RNA-binding domain (DsREB) protein (T2_20487 vs.AT4G00420.2), and MADS-box family protein(T2_22576 vs. AT3G12510.1). To note,NAD6is from the mitochondria genome.
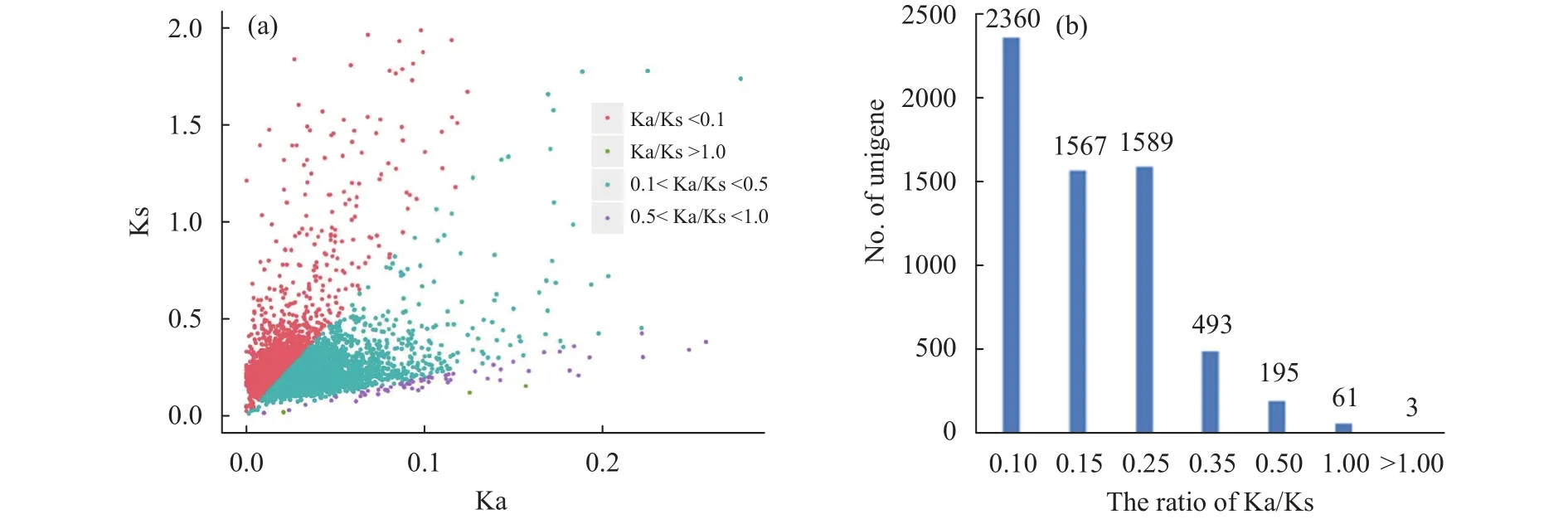
Figure 2 Ka/Ks distribution of 6,268 orthologous gene pairs of B. humilis and A. thaliana. (a) Scatter diagram of Ks and Ka value.The gene pairs with Ks >2 were filtered in this figure and the points with different Ka/Ks ratios are shown in different colour.(b) Histogram of Ka/Ks ratio
The representativeA. thalianagene from each orthologous pairs with a high divergence was subjected to GO annotation and only six genes were assigned into the 'response to stress' GO term. GO enrichment analyses using the Classification SuperViewer revealed that four highly conserved terms were significantly overrepresented in biological process (BP), two in molecular functions (MF), and two in cellular component (CC) categories (p<0.05; Figure 3), while the GO terms 'DNA or RNA metabolism', 'Receptor binding or activity', and 'nucleus' were the most enriched.

Table 1 Positively selected genes in B. humilis
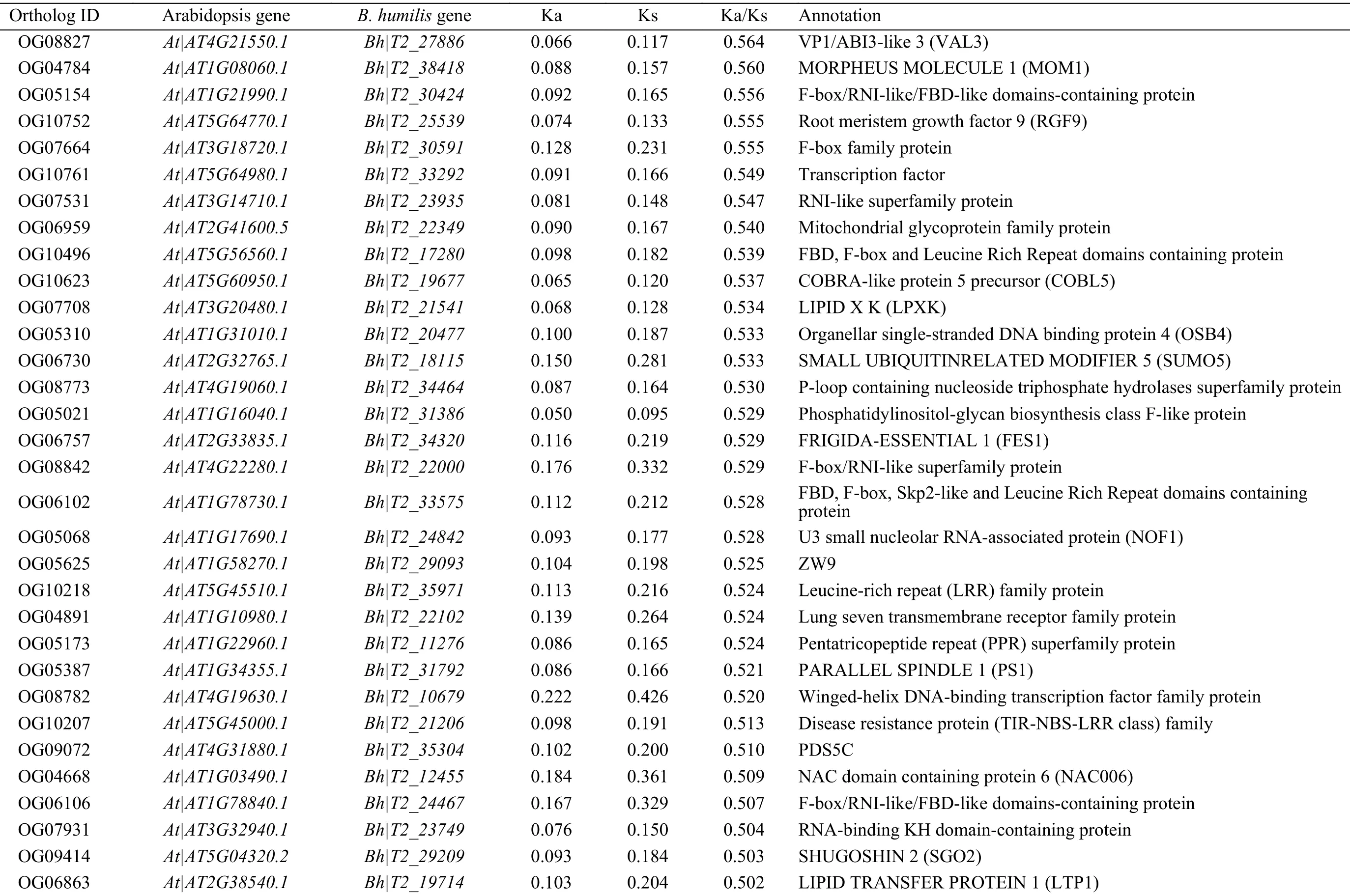
Table 1 Positively selected genes in B. humilis
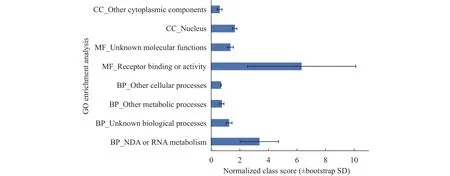
Figure 3 GO enrichment analyses of 64 rapidly evolved genes in B. humilis. The enriched GO terms (p <0.05) were identified using the Classfication SuperViewer Tool and the normalized results are presented. BP, biological process;MF, molecular function; CC, cellular component
4 Discussion
This study identified 64 pairs of genes exhibiting a high divergence betweenB. humilisandA. thaliana.GO annotation revealed that only six genes were categorized into the 'response to stress' BP term and the most enriched GO terms were related to basic biology processes, such as 'DNA or RNA metabolism'.These results are largely consistent with our previous report that the adaptation ofB. humilisto an arid environment is primarily dependent on some of the same genetic components as those inA. thaliana(Zhaoet al., 2017).
4.1 Adaptive value of the DsREB and MADS-box family genes
In this study, three pairs of orthologs were found with strong signs of positive selection.T2_20487was annotated as a DsREB protein. DsREB proteins are essential for the processing and maturation of double-stranded RNA (dsRNA) and function as important regulators of gene expression in response to internal and external stimuli (Hiraguriet al., 2005;Comellaet al., 2008). InA. thaliana, the transcripts ofAT4G00420are specifically overrepresented in shoot apex and female egg cells (Schmidet al., 2005;Winteret al., 2007; Wuestet al., 2010). Although there is no direct empirical evidence to support the function ofAT4G00420, the positive selection signature ofT2_20487may give some hints on the contribution of epigenetic regulation through small RNA pathways to the adaptation ofB. humilisin an arid environment. Another rapidly evolved gene wasT2_22576, which encodes a MADS-box family protein located in the nucleus. The mRNA of itsA. thali-anaorthologue (AT3G12510) is mainly expressed in flower, pollen, guard cell, and seed and is also induced by environmental stresses, such as cold, heat,salt, and osmotic stress (Schmidet al., 2005; Winteret al., 2007; Wanget al., 2008). The protein ofAT3G12510physically interacts with Agamous-like 26 (AGL26,AT5G26880) and AGL47 (AT5G55690)and is involved in the regulation of transcription(Chatr-Aryamontriet al., 2015). In addition, the expression ofAT3G12510is induced by ionizing radiation (Culliganet al., 2006). The finding ofT2_22576under positive selection gives us an indication of howB. humilisadapts to an arid environment.
4.2 Adaptation contribution of the NAD6
It is believed that organelle genes have evolved under strong purifying selection and the organelle DNA variations have been extensively used in phylogenetic and phylogeographic studies during the last decades (Bocket al., 2014). However, the assumption that organelle genome variations are neutral is challenged recently. Physiological studies have shown strong evidence that plant plastids and mitochondria play important roles in orchestrating plant major traits and ecological adaptation (Pastoreet al., 2007; Atkin and Macherel, 2009; Chaveset al., 2009). Based on Ka/Ks ratios, a number of plastome genes under positive selection have been reported to confer plant adaptation to environmental conditions, such as drought stress, CO2concentration and climate variation(Kapralov and Filatov, 2007; Bocket al., 2014). In this study,T2_13757(NAD6) was identified as a positively selected gene. NAD proteins are core components of mitochondria respiratory chain complex I andNAD6is presented in mitochondrial genome of at least 50 angiosperm species (Schertl and Braun, 2014;Sloan, 2015). TheA. thaliana Nad6(ATMG00270)transcripts are over-accumulated after heat stress,combination of drought and heat stress, or cold treatment (Rizhskyet al., 2004; Leeet al., 2005). Mutation of a nuclear-encoded mitochondrial complex I subunitFrostbite1displays reduced cold-responsive gene expression, constitutive accumulation of reactive oxygen species, and impaired cold acclimation(Leeet al., 2002). These lines of evidence suggest that proper mitochondrial respiratory chain complex I is crucial for the plant cold response and maintenance of cellular redox status.
Proline accumulation has long been known as one of the main metabolic responses to environmental stresses in many plant species (Verslues and Sharma,2010). Proline catabolism occurs in mitochondria and is regulated by cellular redox status to sustain plant growth under low water potential condition (Sharmaet al., 2011). A recent study has successfully established the connection among mitochondria genome variation, proline accumulation, and drought resistance (Lovellet al., 2015). Variation inNAD9, a homolog ofNAD6, strongly affects the accumulation of proline via maintaining oxidization/reduction status and in turn regulates plant growth during drought(Lovellet al., 2015). Given the expression pattern ofNAD6and the influence ofNAD9variation on the proline concentration, it is possible thatNAD6functions in a similar mechanism underlying the adaptation ofB. humilisto an arid region.
Genome sequence variations typically exist among populations across the geographic range of a species, which are essential for the local adaptation to the heterogeneous environment (Mitchell-Olds and Schmitt, 2006; Laskyet al., 2014). A comprehensive study on 95A. thaliananatural accessions successfully identified 65 plastid haplotypes and 11 groups of mitochondrial haplotypes and a substantial effect of the cytoplasm genotype on the seed germination efficiency was reported (Moisonet al., 2010). Thus, the distribution of cytoplasmic variations in natural populations should be attributed to natural selection(Greiner and Bock, 2013). Many sequence polymorphisms have been reported inA. thalianamitochondria NADH-dehydrogenase complex genes(Josephet al., 2013) and a high nucleotide substitution has been also observed inNAD6andNAD9sequences of theLupinusgenus (Rureket al., 2003). In addition, two major haplotypes ofA. thaliana NAD9in the 218 accessions show that historical balancing selection has occurred at this locus (Lovellet al.,2015). All these data support the notion that variations of NADH-dehydrogenase complex genes play an important role in plant speciation and local adaptation.
5 Conclusions and perspectives
Comparative analysis is one effective approach to identify the putative genes essential for environment adaptation in the Brassicaceae family. This study presents a comparative evolutionary analysis betweenB. humilisandA. thalianaand a set of 64 gene pairs exhibit a high divergence as revealed by the Ka/Ks ratios. Two nuclear encoded gene pairs and one mitochondria encoded gene pair are under strongly positive selection. These results not only identify some fast evolving nuclear genes, but also highlight the adaptive value of the mitochondria gene during the speciation and adaptation ofB. humilisin an arid environment. Future population genetic analysis of these candidate genes and dissection of the correlations between allele frequencies and climate variables could provide more insights into the adaptive mechanism underlying the wide range adaptation ofB. humilis.
Acknowledgments:
This work was supported by National Natural Science Foundation of China (No. 41201048) and by the Youth Innovation Promotion Association of Chinese Academy of Sciences (2018463).
 Sciences in Cold and Arid Regions2018年5期
Sciences in Cold and Arid Regions2018年5期
- Sciences in Cold and Arid Regions的其它文章
- Study of thermal properties of supraglacial debris and degree-day factors on Lirung Glacier, Nepal
- Comparison of temperature extremes between Zhongshan Station and Great Wall Station in Antarctica
- Numerical simulation of the climate effect of high-altitude lakes on the Tibetan Plateau
- Comparison of precipitation products to observations in Tibet during the rainy season
- Altitude pattern of carbon stocks in desert grasslands of an arid land region
- Comparison of two classification methods to identify grain size fractions of aeolian sediment