
恩施地区腊肠的细菌多样性
2018-10-25 03:29:22王玉荣尚雪娇折米娜葛东颖赵慧君
肉类研究 2018年9期
邓 风,王玉荣,尚雪娇,折米娜,葛东颖,赵慧君,郭 壮,2,*
(1.湖北文理学院食品科学技术学院,鄂西北传统发酵食品研究所,湖北 襄阳 441053;2.恩施市公共检验检测中心,湖北 恩施 445000)
中式腊肠起源于南北朝前期,是一种外形美观、味道鲜醇、营养丰富的传统风味肉制品[1]。传统中式腊肠多采用自然发酵的方式制作,而被称为“世界硒都”的恩施土家苗族自治州境内以山地为主体,少数民族文化多样,较好地保留了这种传统腊肠制作工艺[2]。恩施地区生产的腊肠以猪肉为原料,添加食盐、白酒、八角茴香和花椒等辅料,用松柏木熏制或自然晾晒而成。由于制作环境相对开放,腊肠样品中含有丰富的微生物群系。刘长建等[3]从腊肠中分离出35 株具有降胆固醇能力的乳酸菌,通过对比发现干酪乳杆菌(Lactobacillus casei)清除胆固醇的效率最高。谢科等[4]采用传统分离培养方法从广式腊肠中分离出4 株葡萄球菌(Staphylococcus),使用聚合酶链式反应-变性梯度凝胶电泳(polymerase chain reaction-denatured gradient gel electrophoresis,PCRDGGE)技术分析发现,腐生葡萄球菌(Staphylococcus saprophyticus)、乳杆菌属(Lactobacillus)和木糖葡萄球菌(Staphylococcus xylosus)为其中主要的优势细菌。Wang Xinhui等[5]采用高通量测序技术对中式干腊肠、中式熏腊肠和香肠中的细菌群落结构差异进行分析,发现中式干腊肠、中式熏腊肠中的细菌分布比香肠更为丰富。然而,目前多数研究主要围绕广式腊肠展开,有关恩施地区腊肠微生物多样性的研究仍较少。
大量研究表明,红曲菌[6]、戊糖乳杆菌[7-8]和葡萄球菌[9-10]对腊肠品质的形成具有明显影响,因而广泛开展微生物多样性的解析对腊肠品质的提升具有积极意义。PCR-DGGE[11]和MiSeq高通量测序技术[12]是在发酵食品微生物多样性解析中运用较多的2 项技术,其中PCR-DGGE具有操作简单易行、灵敏度高和可检测到1 个核苷酸水平差异的特点[13],而MiSeq高通量测序技术实现了多样本间微生物多样性的平行比较,具有通量高的优点[14],目前将2 项技术相结合在窖泥[15]、泡菜[16]和豆豉[17]等发酵食品微生物多样性解析中得到了广泛应用。
本研究采用PCR-DGGE与MiSeq高通量测序技术相结合的方法对采集自恩施市的腊肠样品的细菌多样性进行解析,在明确微生物群落结构构成的基础上,为该地区后续腊肠品质的提升提供参考。
1 材料与方法
1.1 材料与试剂
聚丙烯酰胺、冰醋酸、饱和酚、尿素、过硫酸铵、四甲基乙二胺、硝酸银、甲醛、乙二胺四乙酸二钠、N,N-亚甲基二丙烯酰胺和氯仿 国药集团化学试剂有限公司;QIAGEN DNeasy Mericon Food Kit提取试剂盒德国Qiagen公司;Axygen清洁试剂盒 北京科博汇智生物科技发展有限公司;SolutionⅠ、6×Loading Buffer、DNA聚合酶、dNTP mix、pMD18-T Vector和10×PCR Buffer 宝生物工程(大连)有限公司;引物由武汉天一辉远生物科技有限公司合成。
1.2 仪器与设备
HBM-400B拍击式无菌均质器 天津市恒奥科技发展有限公司;DCodeTMSystem 美国Bio-Rad公司;VeritiTM96 孔梯度PCR扩增仪 美国AB公司;5810R台式高速冷冻离心机 德国Eppendorf公司;ND-2000C微量紫外分光光度计 美国Nano Drop公司;Bio-5000 plus扫描仪 上海中晶科技有限公司;MiSeq PE300高通量测序平台 美国Illumina公司;R920机架式服务器美国Dell公司。
1.3 方法
1.3.1 样品采集
分别从湖北省恩施市土桥坝和舞阳坝体育场菜市场((109.47 °N,30.3 °E))采集腊肠样品5 种,编号为LC1~LC5。采集的腊肠样品应符合以下标准:1)腊肠使用猪肉制作而成;2)腊肠无肉眼可见杂质、无霉变且无异味;3)腊肠的制作地需在恩施市行政区域内。
1.3.2 样品预处理及微生物宏基因组DNA提取
将腊肠切碎后,取10 g加入90 mL生理盐水,使用拍击器拍击3 min后,300 r/min条件下离心10 min取上清,上清液10 000 r/min条件下离心10 min后取沉淀,使用QIAGEN DNeasy Mericon Food Kit进行微生物宏基因组DNA提取。
1.3.3 基于DGGE技术的腊肠细菌多样性评价
用灭菌双蒸水将各样品宏基因组DNA的质量浓度调至30 ng/μL,用于后续扩增PCR的扩增体系为25 μL,正向和反向引物分别为LacF-GC-V3F(5’-CG CCCGGGGCGCGCCCCGGGCGGCCCGGGGGCAC CGGGGGCCTACGGGAGGCAGCAG-3’)和Lac-V3R(5’-ATTACCGCGGCTGCTGG-3’)。扩增条件和程序:参照文献[18-19]中的方法,对16S rRNA的V3区进行扩增。扩增结束后用1%的琼脂糖凝胶电泳(2 000 bp的Maker为参照,电压120 V,恒压时间30 min)检测是否扩增出单一、明亮的目的条带[20]。
使用8%的聚丙烯酰胺凝胶,变性范围为35%~52%,将0.5×TAE缓冲溶液的温度调至60 ℃,每个胶孔加入10 μL扩增产物,120 V预电泳78 min,然后80 V固定电压下电泳13 h。电泳结束后的凝胶采用银染法显色,并置于扫描仪上成像[21]。用无菌手术刀回收优势条带,加无菌超纯水过夜,取回溶溶液2 μL进行PCR扩增,扩增使用不带GC夹子的正反引物各0.5 μL以及12.5 μL 2×PCR mix,用无菌超纯水补齐至20 μL[21],扩增程序参照文献[18-19]中的方法。将重新扩增的PCR产物用清洁试剂盒清洁后连接至PMD18-T载体上,然后导入大肠杆菌Top10,将筛选出的阳性克隆送至测序公司测序[22]。
1.3.4 基于MiSeq高通量测序技术的腊肠细菌多样性评价
对样品微生物宏基因组16S rRNA的V3-V4区进行扩增,引物为338F(5’-ACTCCTACGGGAGGCAGCA-3’)和806R(5’-GGACTACHVGGGT-3’),扩增时在引物的5’端加上核酸标签[23],扩增体系和条件参照蔡丽云等[24]的方法。扩增产物检测合格后寄至上海美吉生物医药科技有限公司进行测序,测序平台为Illumian MiSeq PE300。
将测序数据上传至R920机架式服务器端,利用QIIME分析平台[25-26]进行序列分析。原始数据去除低质量序列后采用两步Uclust法[27]在97%的相似度下划分分类操作单元(operational taxonomic units,OTU);从每个OTU中挑选代表性序列与RDP(ribosomal database project,Release 11.5)[28]和Greengenes(Release 13.8)[29]数据库进行比对后,使用FastTree软件[30]构建系统发育进化树,并计算α多样性。
1.4 数据处理
使用多元统计学手段对微生物各分类地位的种类、数量、相对含量及α多样性指数进行计算;序列长度分布图、OTU出现频率和包含序列数统计图以及腊肠中优势核心OTU相对含量的比较分析图用Origin 2017软件绘制;腊肠中优势细菌门属相对含量的比较分析图和腊肠中优势核心OTU相对含量的比较分析图用Excel 2016软件绘制。
2 结果与分析
2.1 基于DGGE技术的腊肠细菌多样性

图 1 腊肠中细菌的DGGE图谱Fig. 1 DGGE fi ngerprint of bacteria in sausage samples
由图1可知,条带6和条带7的亮度远高于其他5 个条带且在每个样品中均存在,说明条带6和条带7所代表的细菌可能为腊肠样品中的优势菌属。条带1~5亮度偏暗,且其仅在某几个腊肠样品中存在,说明这些条带代表的菌属在腊肠中的含量偏低,且可能仅存在于部分样品中。样品LC1的条带数最多,而样品LC5最少,说明样品LC1的细菌多样性最高,样品LC5最低。

表 1 细菌DGGE条带测序结果Table 1 Sequencing of bacterial DGGE bands
由表1可知,经比对发现,腊肠样品中的细菌由Anaerostipes hadrus、约氏不动杆菌(Acinetobacter johnsonii)、Vibrio litoralis、马胃葡萄球菌(Staphylococcus equorum)、热死环丝菌(Brochothrix thermosphacta)和未知分类地位的不可培养细菌构成,且环丝菌属为其优势细菌属。
2.2 基于MiSeq高通量测序技术的腊肠细菌多样性
较之PCR-DGGE技术,以MiSeq为代表的第2代高通量测序技术具有通量高的优点,且实现了菌群的相对定量分析,因而本研究进一步采用MiSeq高通量测序技术对5 个腊肠样品的细菌多样性进行解析,同时对PCR-DGGE的结果进行验证。
通过MiSeq高通量测序,本研究5 个腊肠样品共产生193 486 条高质量的16S rDNA序列,平均每个腊肠样品产生38 697 条,切除引物和Barcode后序列长度的分布情况如图2所示。
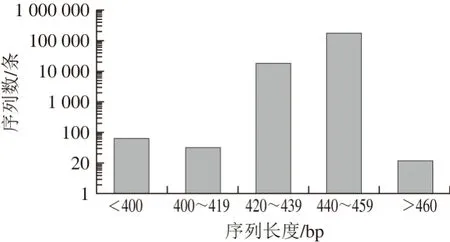
图 2 序列长度分布图Fig. 2 Distribution of sequence length
由图2可知:193 486 条高质量的16S rDNA序列中有175 308 条集中在440~459 bp,占到序列总数的90.61%;18 069 条集中在420~439 bp,占到序列总数的9.34%。使用QIIME平台,对高质量序列进行生物信息学分析,结果表明,共有193 356 条序列通过Align(对齐),按照100%相似性进行Uculst划分后共得到84 979 条代表性序列,按照97%相似性进行Uculst划分后共得到8 057 个OTU,且没有发现嵌合体。
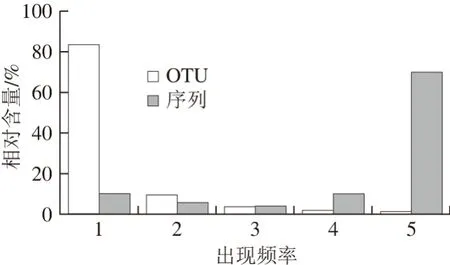
图 3 OTU出现频率和包含序列数统计Fig. 3 Occurrence frequency of OTU and number of sequences within OTU
本研究进一步对OTU在5 个样品中出现的频率和包含序列数进行统计。由图3可知,在5 个样品中出现1、2、3、4 次的OTU分别有6 742、771、289、142 个,分别占OTU总数的83.68%、9.57%、3.59%和1.76%,其包含的序列数分别为18 341、11 093、7 039、19 140 条,分别占所有质控后合格序列数的10.21%、5.76%、3.88%和10.13%。虽然核心OTU仅有113 个,仅占OTU总数的1.40%,但其包含的序列数为137 743 条,占所有质控后合格序列数的70.02%。由此可见,5 个腊肠样品共有大量的细菌类群。
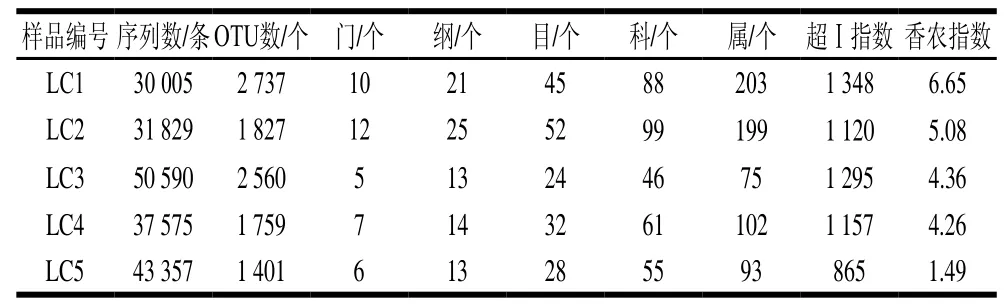
表 2 样品测序结果及各分类地位数量Table 2 Read counts and number of identif i able units at different taxonomical levels
使用RDP和Greengenes数据库比对后,所有序列鉴定为14 个门、58 个纲、90 个目、110 个科和261 个属。由表2可知,样品LC1的超Ⅰ指数和香农指数值均最大,而样品LC5的2 个值均最小,说明样品LC1的细菌多样性最高,而样品LC5最低,与PCR-DGGE结果一致。
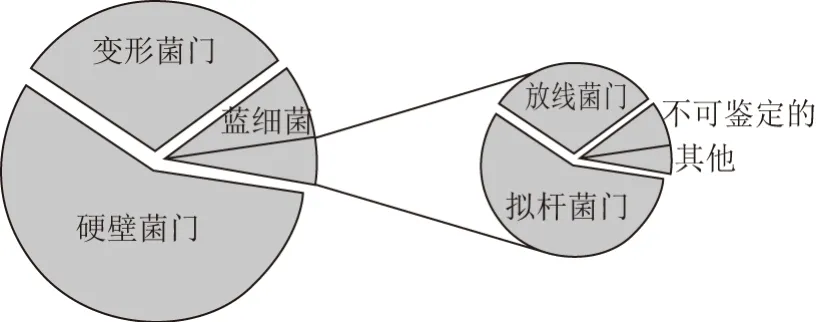
图 4 腊肠中优势细菌门相对含量的比较分析Fig. 4 Comparative analysis of the content of the dominant bacterial phyla in sausage samples
进一步在门水平上对腊肠样品的细菌多样性进行解析。由图4可知,腊肠样品中平均相对含量大于1.0%的细菌门分别为硬壁菌门(Firmicutes)、变形菌门(Proteobacteria)、蓝细菌(Cyanobacteria)、拟杆菌门(Bacteroidetes)和放线菌门(Actinobacteria),其平均相对含量分别为57.01%、30.43%、7.67%、2.63%和2.01%。此外,另有序列被鉴定为TM7、酸杆菌门(Acidobacteria)、梭杆菌门(Fusobacteria)、异常球菌-栖热菌门(Deinococcus-Thermus)、绿弯菌门(Chlorof l exi)、脱铁杆菌门(Deferribacteres)、螺旋体(Spirochaetes)和柔膜菌门(Tenericutes),但其累计平均含量仅为0.13%。由此可见,腊肠样品中的细菌主要隶属于硬壁菌门和变形菌门,其比例占到细菌总数的87.44%。
经RDP和Greengenes数据库比对发现,有13.72%的序列无法鉴定到属水平,相对含量大于1.0%的细菌属如图5所示。
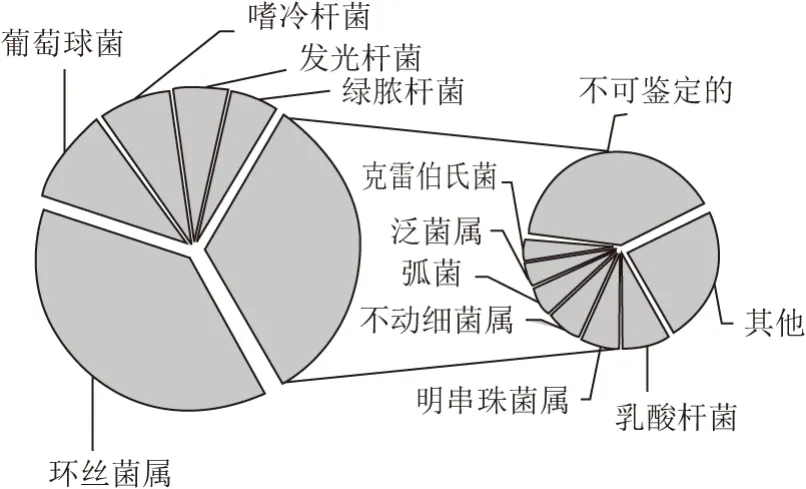
图 5 腊肠中优势细菌属相对含量的比较分析Fig. 5 Comparative analysis of the contents of the dominant bacterial genera in sausage samples
由图5可知,腊肠样品中共有11 个细菌属的平均相对含量大于1.0%,其中环丝菌属(Brochothrix)、葡萄球菌(Staphylococcus)、乳酸杆菌(Lactobacillus)和明串珠菌属(Leuconostoc)4 个属隶属于硬壁菌门,其平均相对含量分别为38.34%、9.79%、2.80%和2.29%。嗜冷杆菌(Psychrobacter)、发光杆菌(Photobacterium)、绿脓杆菌(Pseudomonas)、不动细菌属(Acinetobacter)、弧菌(Vibrio)、泛菌属(Pantoea)和克雷伯氏菌(Klebsiella)7 个属隶属于变形菌门,其平均相对含量分别为7.55%、5.90%、4.82%、2.19%、1.69%、1.48%和1.33%。由此可见,腊肠样品中含量最多的细菌为环丝菌属,与DGGE结果一致。环丝菌属为兼性厌氧的革兰氏阳性杆菌,是导致猪肉在低温冷藏条件下污染的主要细菌之一[31]。此外,葡萄球菌、绿脓杆菌和克雷伯氏菌亦可对食品造成污染[32]。因此,接种乳酸杆菌和明串珠菌属进行腊肠的纯种发酵,在保持腊肠加工和贮藏环境清洁的同时,对提升腊肠相关产品的安全品质可能具有积极意义[7]。
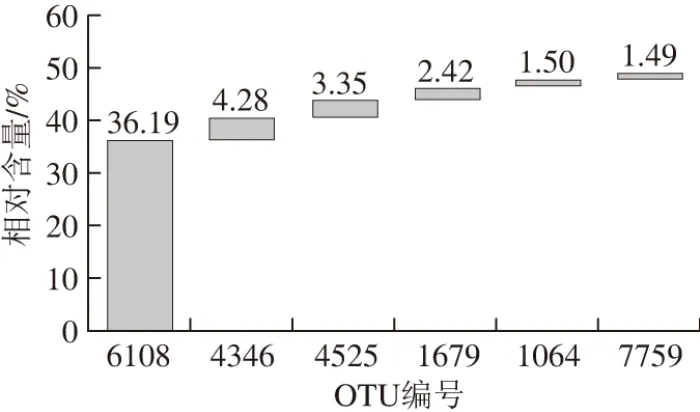
图 6 腊肠中优势核心OTU相对含量的比较分析Fig. 6 Comparative analysis of the contents of the dominant core OTUs in sausage samples
对142 个相对含量大于1.0%的核心OTU进行统计和分析。由图6可知,腊肠样品中OTU 6108(隶属于Brochothrix)、OTU 4346(隶属于Psychrobacter)、OTU 4525(隶属于Staphylococcus)、OTU 1679(隶属于Lactobacillus)、OTU 1064(隶属于Vibrio)和OTU 7759(隶属于Staphylococcus)的平均相对含量分别为36.19%、4.28%、3.35%、2.42%、1.50%和1.49%,均大于1.0%。由此可见,5 个腊肠样品共有大量的细菌类群,6 个核心OTU占到了序列总数的49.23%。
3 结 论
MiSeq高通量测序结果表明:恩施地区腊肠样品中的细菌主要隶属于硬壁菌门和变形菌门,二者比例占到细菌总数的87.44%;113 个核心OTU中的序列占所有质控后合格序列数的70.02%,说明5 个腊肠样品共有大量的细菌类群。PCR-DGGE与Illumina MiSeq 检测结果均表明恩施腊肠中优势细菌属为环丝菌属。
猜你喜欢
国际太空(2023年1期)2023-02-27 09:03:42
少年文艺·开心阅读作文(2022年2期)2022-01-10 09:19:52
作文周刊·小学一年级版(2020年48期)2020-12-15 10:51:19
透析与人工器官(2020年1期)2020-11-16 01:42:34
中国比较医学杂志(2020年4期)2020-05-26 05:47:22
中成药(2019年12期)2020-01-04 02:02:40
铁道通信信号(2019年8期)2019-10-10 05:06:00
水生生物学报(2019年4期)2019-07-20 08:08:10
生物安全学报(2019年3期)2019-02-15 16:54:12
川北医学院学报(2019年6期)2019-02-10 10:48:32
