
甘精胰岛素肽图分析方法研究
2018-10-23 11:27:42王绿音杨慧敏李晶张慧李懿张伟梁成罡
生物技术通讯 2018年5期
王绿音,杨慧敏,李晶,张慧,李懿,张伟,梁成罡,
1.中国食品药品检定研究院,北京 102629;2.中国药科大学 生命科学与技术学院,江苏 南京 210009
甘精胰岛素是由重组DNA技术表达的具有长效作用的人胰岛素类似物,适用于Ⅰ、Ⅱ型糖尿病的治疗[1]。甘精胰岛素由2条肽链组成,相对分子质量为6063.0。与人胰岛素相比,它在结构上发生了如下2个变化:首先,用甘氨酸取代了A链21位的天冬酰胺;其次,在B链的C端添加了2个带正电荷的精氨酸[2]。这些改变将甘精胰岛素的等电点由人胰岛素的5.4升高至6.7,皮下注射甘精胰岛素后,在生理pH值条件下甘精胰岛素的溶解度下降,分子聚集成六聚体形成药物沉淀,缓慢持续地释放少量甘精胰岛素,从而产生长效、平稳、无峰值的血药浓度,有效控制空腹血糖,降低低血糖风险[3-4]。
肽图分析是验证生物技术产品一级结构完整性及正确性最有效的方法之一,同时也是考察产品的批间一致性及其工程菌遗传稳定性的重要手段。该项目系生物技术产品的常规检测指标,对产品的质量控制有非常重要的意义[5]。V8蛋白酶是一种可特异性作用于谷氨酸C端的丝氨酸蛋白酶,在适宜条件下可特异性切割与甘精胰岛素A链第4、17位,B链第13、21位的谷氨酸残基C端相结合的肽键。甘精胰岛素A、B链之间由2对二硫键连接,故水解后产生4个理论肽段(Ⅰ、Ⅱ、Ⅲ及Ⅳ)[6-8](图1)。目前各国药典及国内外企业均采用V8酶水解甘精胰岛素,然后用RPHPLC分离4个目标肽段得到其肽图谱[9-10]。研究中发现,采用《欧洲药典》9.5版(European Pharma⁃copoeia 9.5,EP9.5)甘精胰岛素肽图分析方法中的酶解条件,反应的完全性及有效性不佳,从酶解后典型的色谱图来看,主成分消化不完全,非特异酶解肽段含量较高,理论目标肽段中的Ⅳ和Ⅰ含量过低,有时甚至无法有效检出,无法确保除酶切位点外的其他序列的正确性及完整性[9],不适于推广为行业标准方法。近年来各申报单位均舍弃了上述方法,以中国药典2015年版(Chp2015)重组人胰岛素肽图分析方法[11]为基础进行甘精胰岛素肽图分析,但在酶解反应的完全性、酶解产生的理论目标肽段的稳定性等方面仍然存在问题。因此,为满足甘精胰岛素日常质控和科学监管及评价需求,研究建立酶解反应完全、特异性强、肽段分离效果好的甘精胰岛素肽图分析方法,具有充分的必要性和现实紧迫性。
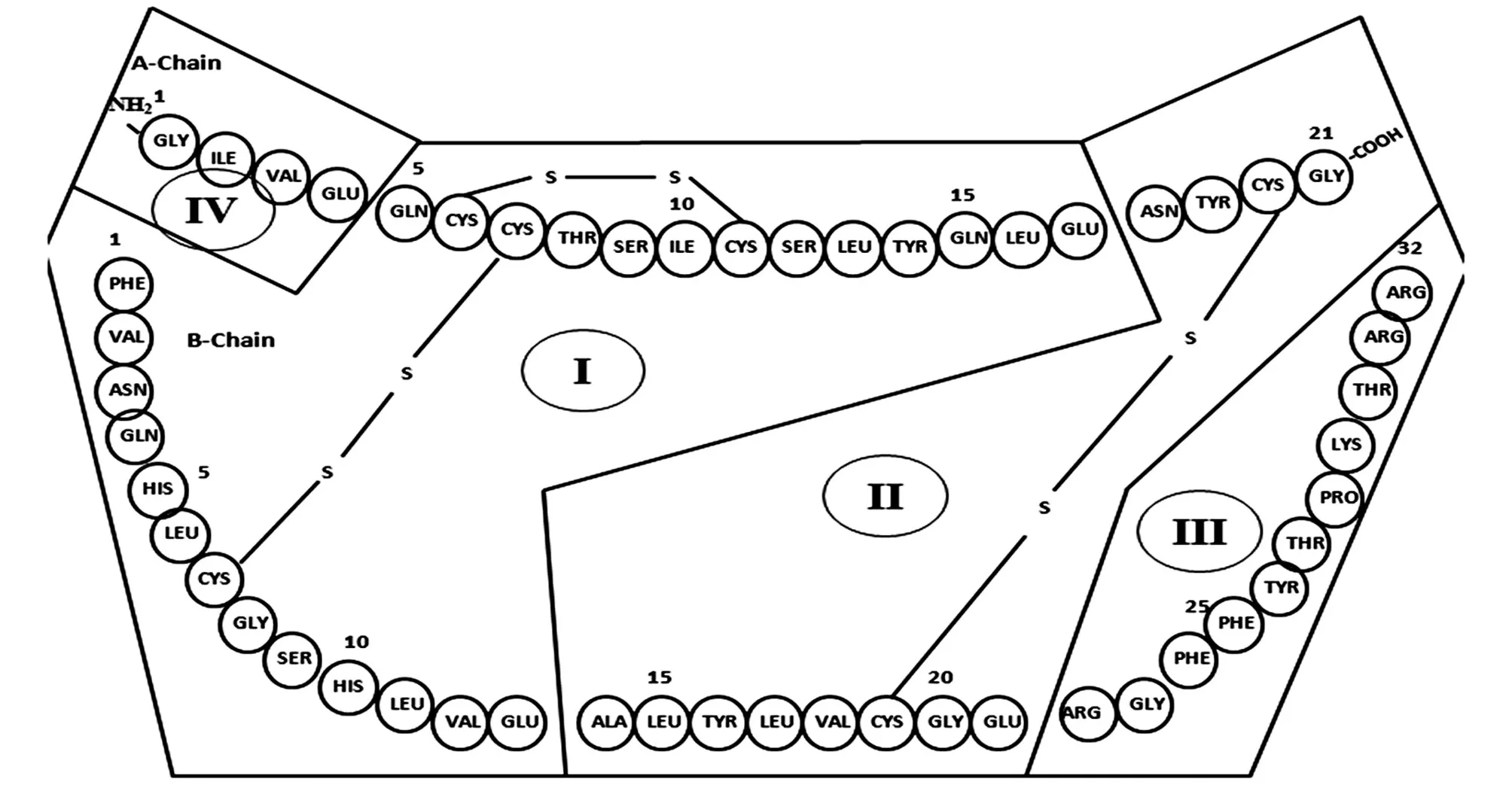
图1 甘精胰岛素结构图及V8蛋白酶切位点
我们通过优化酶解条件中的盐浓度及pH值、酶量、反应温度与时间等关键因素,大大改善了甘精胰岛素的酶解效果,建立了满足上述要求的肽图分析方法,根据Chp2015四部通则9101[12]药品质量标准分析方法验证指导原则进行了专属性、中间精密度、重复性及耐用性考察,并通过LC-MS对各肽段的色谱峰进行了准确鉴别。为甘精胰岛素产品的研发生产与申报提供技术支持及质控标准参考,也为新版中国药典该品种肽图鉴别质量标准的制订与提高打下基础。
1 材料和方法
1.1 材料
甘精胰岛素样品来自国内外不同生产企业;甘精胰岛素国家对照品(批号140701-20040)及B32脱精氨酸甘精胰岛素对照品(410017-201701)均来自中国食品药品检定研究院;V8酶(1000 U/mg,1 mg/支,Worthington公司);盐酸、Tris、磷酸、硫酸钠(分析纯,国药集团化学试剂有限公司);三氟乙酸(纯度≥99.5% ,Alfa Aesar公司);乙腈(色谱纯,Fisher Scientific公司)。
精密称取56.804 g硫酸钠,溶于800 mL纯水,磷酸调节pH2.3,加水定容至1 L,即为0.2 mol/L硫酸盐缓冲液。精密称取4.8456 g Tris,溶于90 mL纯水,稀盐酸调节pH9.0,加水定容至100 mL,即为0.4 mol/L Tris-HCl(pH9.0)缓冲液。吸取900 μL稀盐酸于800 mL超纯水中,定容至1 L,即为0.01 mol/L盐酸溶液。取V8酶,加水制成5000 U/mL的溶液,分装至0.5 mL离心管中,-20℃保存,用前取出。
LC-20A高效液相色谱仪(包括Prominence LC-20AD/20AT二元泵、Prominence SIL-20A自动进样器、Prominence SPD-20A/20AV UV-VIS检测器、LC-solution色谱工作站,岛津公司);Acquity UPLC超高效液相色谱-Xevo G2 Q Tof串联四级杆飞行时间质谱仪(Waters公司);HAAKE-S30不锈钢加热水浴循环器(Thermo Scientific公司);ME155DU电子天平(Mettler Toledo公司)。
1.2 甘精胰岛素肽图分析方法的建立
1.2.1 样品处理 分别取甘精胰岛素样品及对照品原料适量,各加0.01 mol/L盐酸溶液溶解并稀释至浓度为10 mg/mL的溶液。取20 μL 10 mg/mL的甘精胰岛素样品及对照品溶液,加至98 μL 0.4 mol/L Tris-HCl(pH9.0)溶液中,加入 V8酶溶液(5000 U/mL)4 μL,纯水78 μL,混匀,置37℃水浴中保温1 h,取出后加入3 μL磷酸终止反应,混匀,即为供试品及对照品溶液,4℃保存。
1.2.2 色谱条件 采用Waters Symmetry 300 C18色谱柱(150 mm×4.6 mm,3.5 μm),以0.2 mol/L硫酸盐缓冲液(pH2.3)-乙腈(90∶10)为流动相A、乙腈-水(50∶50)为流动相B,梯度洗脱(0~5 min,90% ~80% A;5~45 min,80% ~40% A;45~50 min,40% A),流速1 mL/min,检测波长214 nm,柱温40℃,进样量20 μL。
1.2.3 LC-MS条件 按上述方法处理样品,对酶解产生的各肽段进行液相色谱分离并收集,分别进行LC-MS分析。
1.2.3.1 超高效液相色谱条件 采用Waters Ac⁃quity UPLC Peptide CSH C18色谱柱(100 mm×2.1 mm,1.7 μm),以0.1% 甲酸水溶液为流动相A、0.1% 甲酸乙腈溶液为流动相B,梯度洗脱(0~6.95 min,95% ~60% A;6.95~7.29 min,60% ~95% A;7.29~10.50 min,95% A),流速0.3 mL/min,检测波长214 nm,柱温40℃,进样量10 μL。
1.2.3.2 质谱条件 采用电喷雾离子源(ESI),正离子检测模式,脱溶剂器温度450℃,源温80℃,毛细管电压3.0 kV,cone电压40 V,collision电压Low CE 6eV,collision电压High CE 6eV,数据采集范围m/z为100~2000,采用MSE模式获得各肽段母离子一级及二级质谱数据。
1.2.4 数据分析 采用LC-solution色谱工作站对样品的理论肽段进行积分,记录肽段Ⅱ和Ⅲ的分离度及拖尾因子。通过计算各肽段的相对保留时间RRT(样品肽段的保留时间与对照品肽段的保留时间的比率)对甘精胰岛素进行阳性鉴别。
用Masslynx 4.1软件采集数据,用Biophaml⁃ynx 1.3.2软件进行数据处理。选择V8酶作为酶切试剂,选择30 ppm的质量偏差,漏切位点数设为1,选择N端焦谷氨酸环化为固定修饰。
1.3 甘精胰岛素肽图分析方法的优化
对缓冲体系中的Tris浓度与pH值、酶量及酶解温度与时间进行了优化。选择0.2 mol/L Tris-HCl(pH9.0)、0.4 mol/L Tris-HCl(pH8.0)及 0.4 mol/L Tris-HCl(pH9.0)作为 3 个候选酶解缓冲液;酶量的考察,分别加入 2 μL(10 U)、4 μL(20 U)、6 μL(30 U)V8酶水解200 μg甘精胰岛素;选择45℃水浴1 h、37℃水浴0.5 h、37℃水浴1 h及37℃水浴2 h作为4个候选的酶解温度与时间条件。所有实验均遵循改变其中1个条件其余保持不变的原则,取同一份样品按上述不同条件下的制备方法处理后进样分析。
1.4 所建方法与现有方法的对比
方法1:按1.2.1项样品处理方法及1.2.2项色谱条件测定甘精胰岛素肽图;方法2:按EP9.5甘精胰岛素肽图鉴别项下的样品处理方法及1.2.2项色谱条件测定甘精胰岛素肽图;方法3:按Chp2015版重组人胰岛素肽图鉴别项下的样品处理方法及1.2.2项色谱条件测定甘精胰岛素肽图。
1.5 方法学验证
1.5.1 专属性 取0.01 mol/L盐酸溶液代替样品按上述方法处理,作为V8酶对照;取20 μL 10 mg/mL的甘精胰岛素样品,加至78 μL酶解缓冲液中,加入82 μL纯水,水浴后终止反应,作为甘精胰岛素样品对照;另取B32脱精氨酸甘精胰岛素对照品,按上述方法加入V8酶处理,作为B32脱精氨酸甘精胰岛素样品对照。所有酶解后的样品均按上述色谱条件进行肽图分析。
1.5.2 重复性(精密度) 取对照品溶液,按1.2.1项制备甘精胰岛素样品,消化后的溶液按1.2.2项色谱条件重复进样6次,考察甘精胰岛素4个特征肽段保留时间和峰面积的相对标准偏差(RSD)。
1.5.3 中间精密度 由2名实验者于同一时间按1.2.1方法制备2份样品,由同一实验人员于不同时间同法制备3份样品,每份样品进样2次分析。
1.5.4 耐用性 取甘精胰岛素对照品,按1.2.1项处理,分别加入V8酶15、20及25 U,得到加入不同酶量后的样品消化溶液。对甘精胰岛素对照品按1.2.1及1.2.2的方法酶解后分析,设置柱温为38℃、40℃、42℃,得到不同柱温下的样品肽图谱。此外,采用Waters Symmetry300、Aglient ZOR⁃BAX SB及Phenomenex Luna C18色谱柱对同一份样品消化液进行肽图分析。
1.5.5 酶解后样品的稳定性 酶解后的甘精胰岛素样品存放于HPLC仪器的样品室(6℃)中,于0、12、24 h各进样分析。将酶解后的样品分装后存放于-20℃,于0、1、7 d时,取出溶解并进样分析。
2 结果
2.1 甘精胰岛素肽图分析方法的建立
按1.2项下的方法分析甘精胰岛素的肽图,HPLC结果见图2,色谱图中主要包括4个理论肽段及3个非特异酶解肽段。通过与未处理的甘精胰岛素样品色谱图比较,可确定肽段Ⅵ为未消化的主成分。其余2个非特异酶解肽段则被推测为位于肽段Ⅰ之后的Ⅳ+Ⅰ片段[2]及肽段Ⅰ′,通过下述LC-MS进行鉴定。肽段Ⅱ和肽段Ⅲ的分离度大于25.0,且二者的拖尾因子均小于1.5,理论目标肽段Ⅳ、Ⅲ、Ⅱ、Ⅰ的RRT均在1.00±0.01范围内。LC-MS分析得到的各肽段一级质谱结果见表1、图3,肽段Ⅳ、Ⅲ、Ⅱ、Ⅰ的实测离子质量与理论值一致。通过MSE高能量碎裂获得的二级碎片b、y离子信息,确定了4个目的肽段氨基酸序列的正确性,证实了位于肽段Ⅰ之后的2个非特异酶解肽段分别为肽段Ⅳ+Ⅰ及Ⅰ′(图4),该结果也与我们的推测一致。
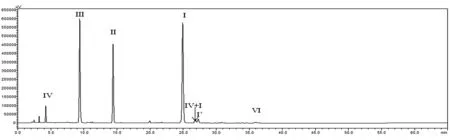
图2 新建方法测得的甘精胰岛素肽图谱
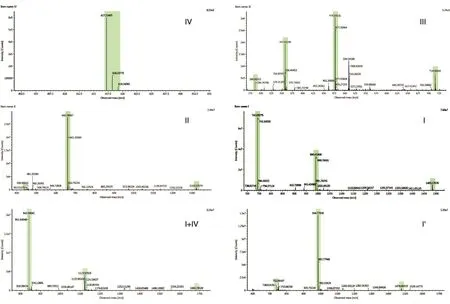
图3 甘精胰岛素肽段一级质谱图

图4 甘精胰岛素Ⅰ+Ⅳ肽段、Ⅰ′肽段MSE谱图
研究表明,肽段Ⅰ′在肽段Ⅰ之后出峰,是酶解过程中由肽段Ⅰ的A链N端第5位谷氨酰胺自身环化形成的焦谷氨酸衍生物Glp(A5)-I[8]。该衍生物来源于肽段Ⅰ,其生成量与环境温度、pH值密切相关[13-14],本研究以肽段Ⅰ′与Ⅰ的峰面积之比作为衡量肽段Ⅰ稳定性的指标。计算图2中Ⅰ′与Ⅰ的峰面积比值为0.05% ,表明采用本方法酶解所得的肽段Ⅰ的稳定性较好。
以未消化的主成分比率(肽段Ⅵ与较为稳定的肽段Ⅱ的峰面积之比)作为衡量酶解完全性的指标。计算图2中肽段Ⅵ与Ⅱ的峰面积比值为0.05% ,表明采用本方法测得的未消化的主成分含量较低,酶解的完全性较好。
2.2 甘精胰岛素肽图分析方法的优化
对酶解条件中的几个关键参数进行了优化,以不同条件得到的非特异酶解肽段(未消化的主成分、片段Ⅳ+Ⅰ和片段Ⅰ′)的含量作为判定候选条件优劣的指标。由图5可见,采用0.4 mol/L Tris-HCl(pH9.0)作为酶解缓冲液处理样品,非特异酶解肽段的含量最低,酶解最为完全。
当体系中加入 V8酶的量为 4 μL(20 U)时(图6),主成分的酶解程度优于2 μL(10 U),且与加入6 μL(30 U)时的酶解程度相当。低酶量(10 U)导致的不完全酶解使得肽段Ⅵ+Ⅰ的含量显著高于20~30 U酶量条件下该肽段的含量。此外,由于酶量增多,酶解更加完全,加入6 μL(30 U)V8酶产生的肽段Ⅵ+Ⅰ的量略低于4 μL(20 U)条件,但二者相差不大,故选择4 μL(20 U)作为最佳酶量。
在4个酶解温度与时间条件中,37℃反应0.5 h及45℃反应1 h得到的片段Ⅳ+Ⅰ的量明显高于于另外2个条件。此外,37℃反应2 h所得的片段Ⅳ+Ⅰ的量略低于37℃反应1 h,这是因为随着反应时间的延长,片段Ⅳ+Ⅰ被酶切得更加充分。然而随着酶解反应时间的延长,水解衍生物焦谷氨酸(Ⅰ′)增多,综合实验效率及酶解效果,选择37℃水浴1 h为最佳反应条件(图7)。

表1 甘精胰岛素主要肽段及离子化信息
2.3 所建方法与现有方法的对比
比较采用所建方法、EP9.5甘精胰岛素肽图分析法、Chp2015重组人胰岛素肽图分析法中的酶解条件处理样品,并按照1.2.2项色谱图条件测定的肽图。Chp(图8A)及本方法(图8C)的色谱图可清晰地呈现4个理论肽段,而EP9.5方法的色谱图(图8B)中肽段Ⅰ和Ⅳ无法被有效检出。
在酶解反应的完全性方面,计算3种方法测定的未消化的主成分比率(肽段Ⅵ与Ⅱ的峰面积之比),其中本方法(图8C)的未消化主成分比率为0.05% ,远低于Chp(图8A)的0.23% 及EP9.5(图8B)的1.14% ,表明3种方法中,Chp方法的酶解效率高于EP9.5,而新方法的酶解反应效率最高,酶解完全性更好。3种方法中肽段Ⅵ+Ⅰ的含量差异也很好地佐证了上述结论,其中EP9.5方法中肽段Ⅳ+Ⅰ的含量更是高至与主要肽段Ⅲ相当。
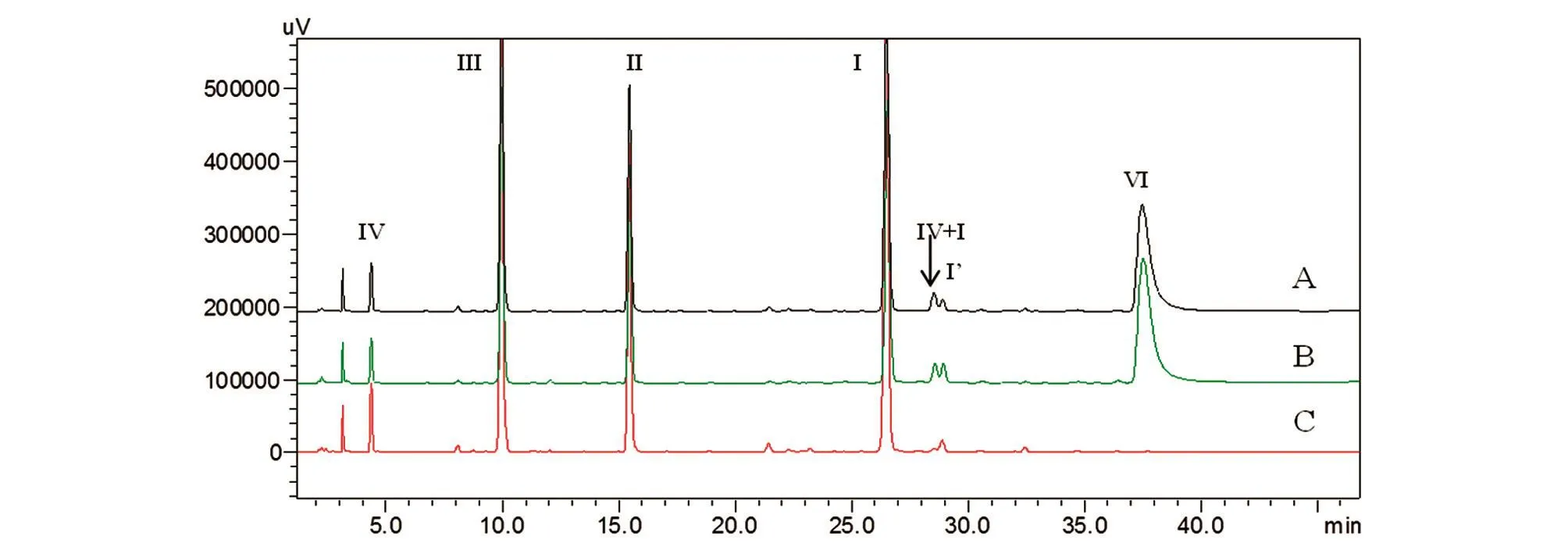
图5 用不同酶解缓冲液酶解后的甘精胰岛素肽图谱
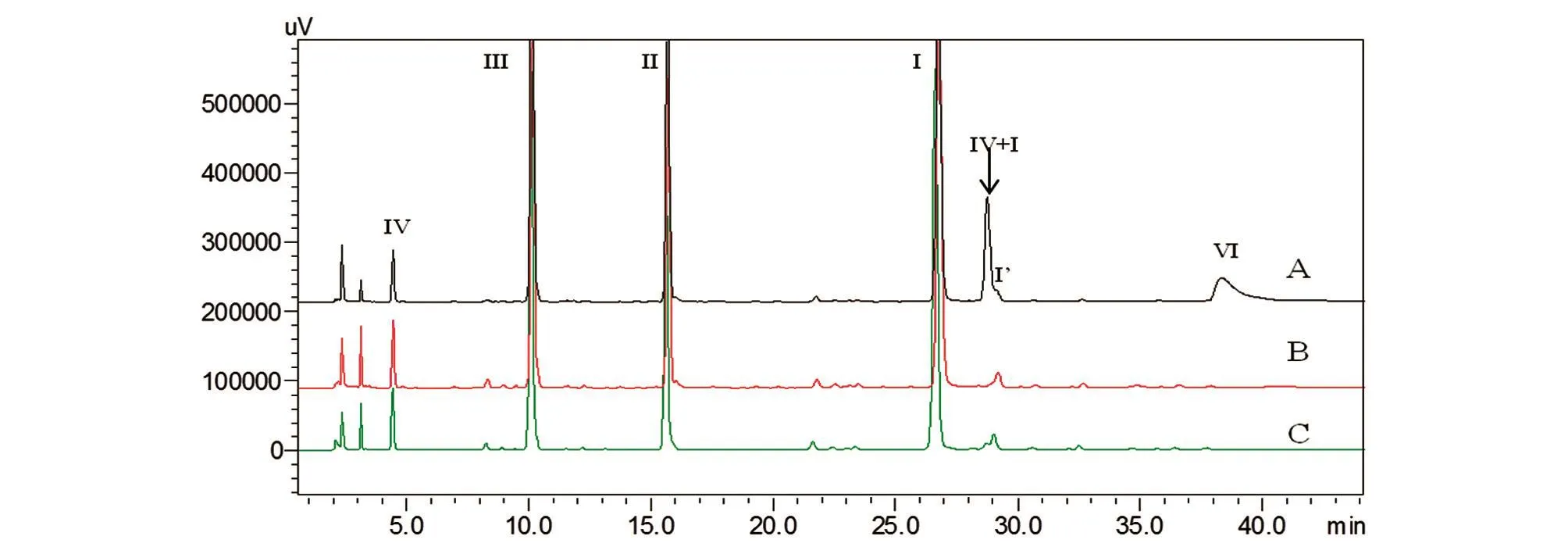
图6 用不同酶量酶解后的甘精胰岛素肽图谱
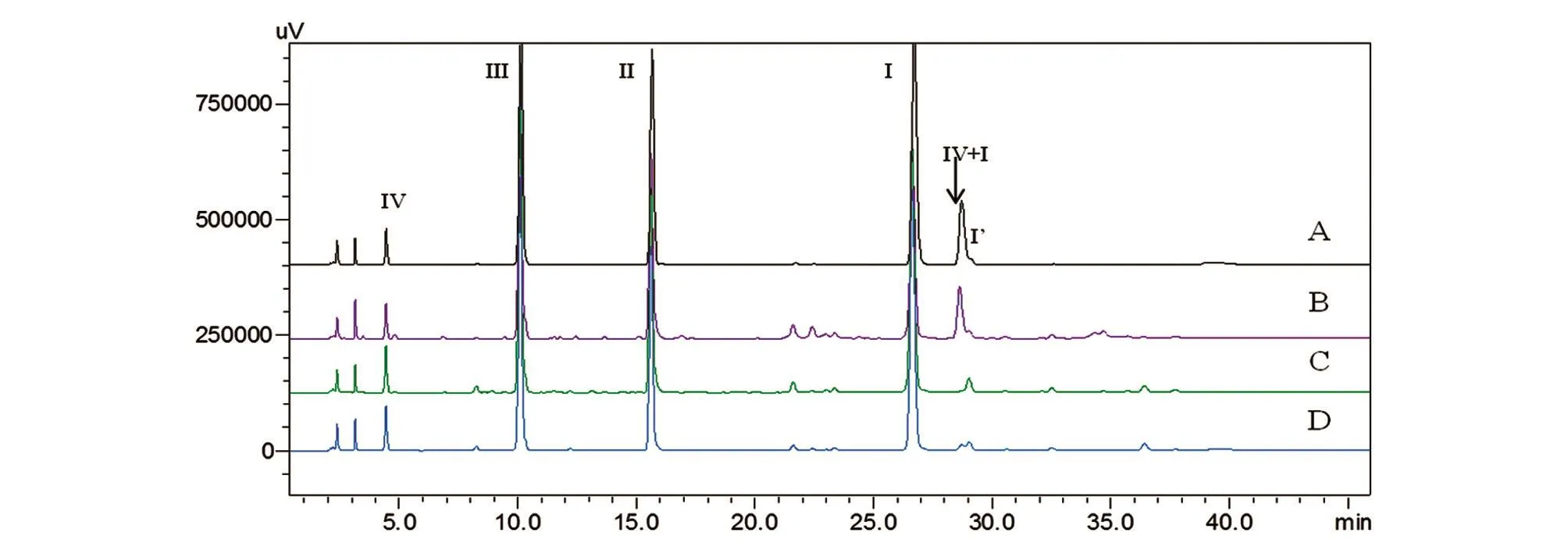
图7 用不同酶解温度和时间酶解后的甘精胰岛素肽图谱
在酶解产生肽段的稳定性方面,考察3种方法所得肽段Ⅰ的稳定性,计算Ⅰ′与Ⅰ的峰面积比值,本方法(图8C)为0.05% ,远低于Chp(图8A)的0.09% ,表明本方法肽段Ⅰ的稳定性更好。
2.4 方法学验证
2.4.1 专属性 专属性考察结果表明V8酶对照在样品各肽段出峰时间均无干扰峰出现(图9E)。B32脱精氨酸甘精胰岛素肽图谱中的肽段Ⅲ′与甘精胰岛素的肽段Ⅲ相比,仅在B链C端缺失一个亲水的精氨酸,导致肽段Ⅲ′疏水性增强,出峰时间明显晚于甘精胰岛素的肽段Ⅲ(图9B),2种胰岛素对照样品的结果也与之对应(图9C、D)。这些结果表明本方法分辨率较高,可精准反映甘精胰岛素一级结构的正确性,专属性较好。
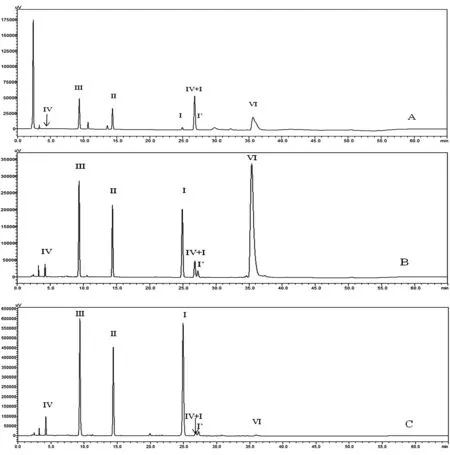
图8 3种酶解方法测得的甘精胰岛素肽图谱
2.4.2 重复性(精密度) 考察甘精胰岛素4个特征肽段保留时间和峰面积的RSD,由表2可见,各肽段保留时间及峰面积的RSD均≤0.20% ,表明方法的精密度较好,符合肽图分析的技术要求。
2.4.3 中间精密度 以样品中各肽段的相对保留时间(色谱图上样品特征峰的保留时间与对照品对应峰的保留时间之比)的RSD作为评价中间精密度的指标,结果见表3。各肽段相对保留时间均在1.00±0.01范围内,RSD为0.12% ~0.26% ,表明不同人员及不同时间下制备样品测得结果的变异程度较小,方法的中间精密度良好。
2.4.4 耐用性 耐用性考察结果见表4,当反应体系中的酶量与色谱条件中的柱温发生微小变动时,各肽段相对保留时间均在1.00±0.01范围内,RSD为0.01% ~0.36% 。此外,对同一份样品采用不同品牌的色谱柱测定,各肽段的相对保留时间RSD均小于0.35% ,肽段Ⅲ与Ⅱ的拖尾因子均小于1.3,分离度均大于25.0(图6)。这些结果表明上述测定条件微小变化时,测定结果受影响的程度不大,方法的变异度低,耐用性较好。
2.4.5 酶解后样品的稳定性 考察了样品的储存稳定性(6℃)及冻存(-20℃)稳定性。由表5可见,样品消化液于6℃存放24 h及-20℃条件下储存7 d后,测定结果依然较稳定。

表2 重复性(精密度)考察结果(n=6)

表3 中间精密度考察结果(n=6)
3 讨论
肽图分析作为控制基因重组药物一致性的重要手段,对重组蛋白的结构确证和专属性鉴别具有重要意义[15-17]。该方法常通过酶解蛋白质产生肽段碎片,然后对碎片进行可重现的分离及鉴定,包括蛋白质前处理、酶解、特征肽段的分离及鉴别这3个主要步骤。一个好的肽图分析方法,除须具备高度的特异性、可鉴定酶切位点处序列信息的正确性以外,还应具备酶解反应完全、理论目标肽段分离度良好且清晰可辨、理论目标肽段在酶解体系中稳定性较好、非特异酶解肽段的含量较低等特点,从而实现对蛋白质中单一氨基酸的氧化、脱氨等变化的精确检测。
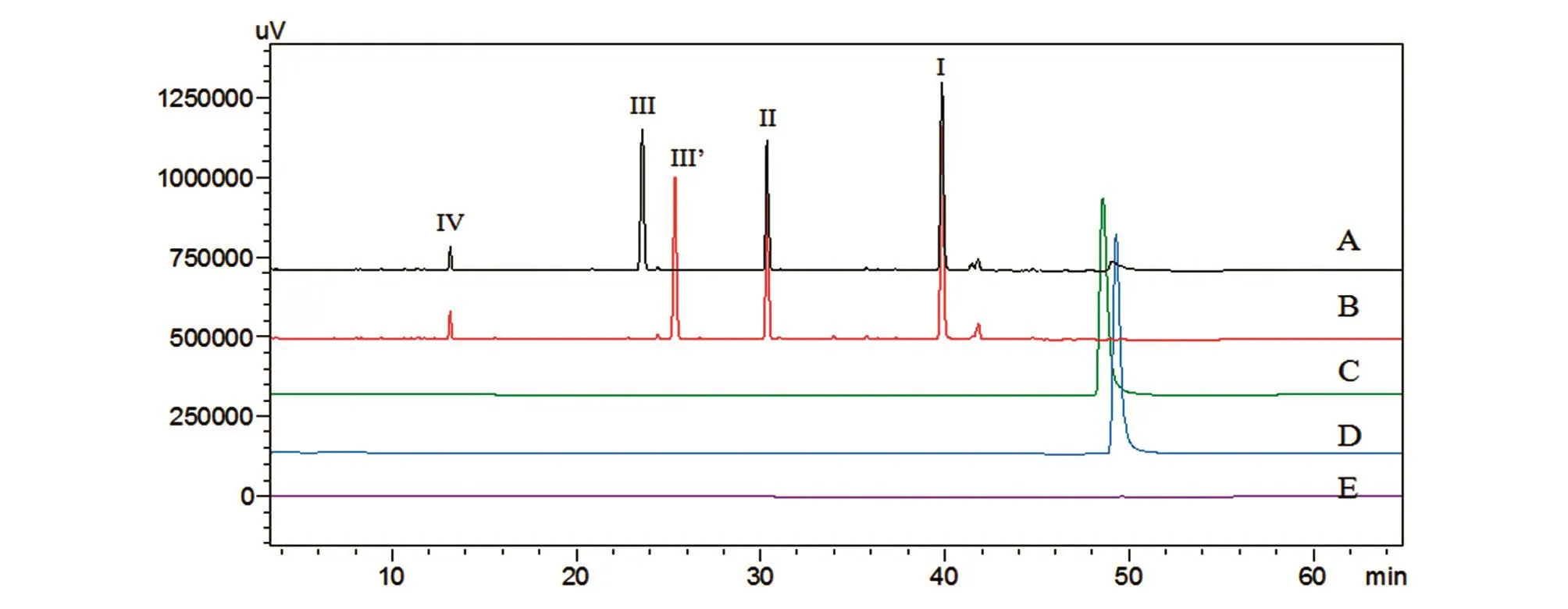
图9 专属性色谱图
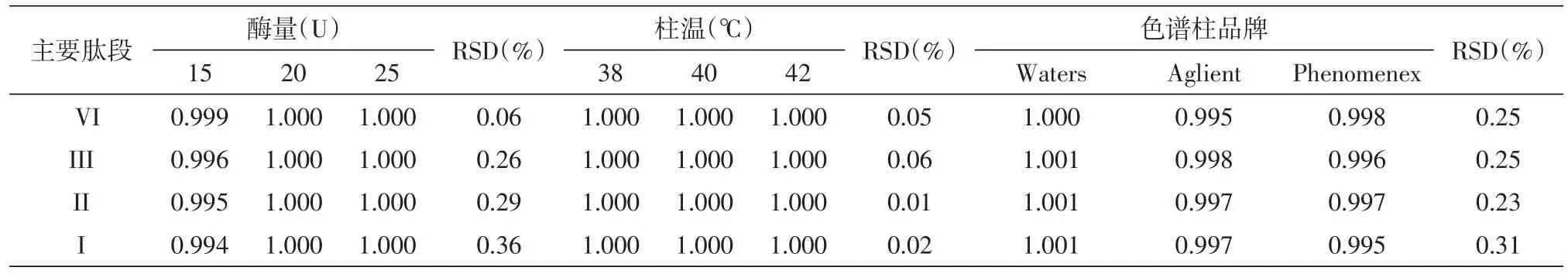
表4 耐用性考察结果(n=3)
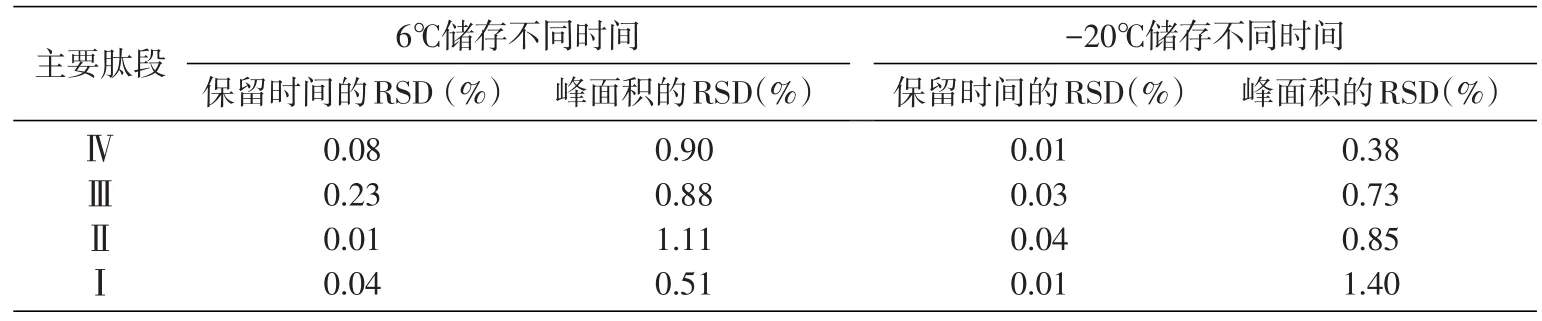
表5 酶解后样品的稳定性考察结果(n=3)
现行的甘精胰岛素肽图分析药典方法收载于EP9.5,该方法在酶解的完全性及理论目标肽段的检出等方面存在问题,因此各申报单位纷纷参考Chp2015重组人胰岛素肽图分析方法。该方法适用于等电点为5.4的重组人胰岛素,对于等电点为6.2的甘精胰岛素而言,仍存在样品无法完全溶解、非特异酶解肽段含量较高等缺陷。研究中发现,提高缓冲体系中的盐浓度与pH值,可增加甘精胰岛素样品的溶解度,从而在一定程度上降低未消化的主成分的含量。此外,温度越高、pH值越低,酶解过程中产生的焦谷氨酸衍生物也会增多。因此,充分考察并对这些影响酶解效果的关键因素进行优化,对于甘精胰岛素肽图分析方法的建立至关重要。
本研究对酶解条件中的关键因素进行了优化,包括缓冲体系中的盐浓度和pH值、V8蛋白酶的量、酶促反应温度与时间,最终确定了最佳酶解条件。在该条件下,酶解反应更充分,甘精胰岛素完整蛋白基本被完全酶解,非特异酶解肽段产生较少,较大程度地降低了其对表征理论目标肽段的干扰。比较采用本方法与EP9.5甘精胰岛素、Chp2015重组人胰岛素肽图分析方法的酶解条件测得的肽图谱可见,本方法在酶解反应的完全性、特异性、酶解产生肽段的稳定性等方面具有显著优势。通过LC-MS法对酶解产生的各目标肽段进行了确证,并且首次对2个酶解产生的非特异肽段(Ⅰ+Ⅳ及Ⅰ′)进行了鉴别。方法学验证结果表明,该方法对甘精胰岛素有很好的专属性,重复性试验的RSD在0.20% 以内,中间精密度试验测得的相对保留时间RSD为0.01% ~0.36% 。此外,酶量及柱温的微调、更换不同品牌的色谱柱等并未给实验结果带来明显变化,表现了良好的耐用性。且酶解后样品溶液的储存稳定性及冻存稳定性均较好。以上数据显示该方法适于相应检测要求,具有相当的准确性和可靠性,可以达到控制产品质量的目的。
综上,我们建立了良好的甘精胰岛素肽图分析方法,该方法可作为甘精胰岛素结构确证与一致性比较的常规检验方法,并广泛用于工艺开发、过程控制与终产品的放行检验。本研究为该品种质量标准的提高打下了基础,同时也可为其他胰岛素类产品的肽图鉴别提供参考。
猜你喜欢
北京航空航天大学学报(2021年9期)2021-11-02 08:24:32
World Journal of Clinical Cases(2020年17期)2020-09-18 08:03:24
重型机械(2020年2期)2020-07-24 08:16:16
检验医学与临床(2020年1期)2020-01-10 04:44:22
中国航海(2019年2期)2019-07-24 08:26:40
中国医药指南(2017年3期)2017-11-13 02:55:26
中国卫生标准管理(2015年16期)2016-01-20 09:26:24
应用海洋学学报(2015年2期)2015-11-22 07:36:40
中国当代医药(2015年32期)2015-03-01 02:08:51
现代检验医学杂志(2015年6期)2015-02-06 01:44:25
