
荧光碳点的合成及其在绿麦隆检测中的应用研究
2018-10-16 03:21廖秀芬曾秋莲易忠胜韦红玲梁杨琳欧玉玲吕春秋
分析测试学报 2018年9期
廖秀芬,曾秋莲,易忠胜,韦红玲,梁杨琳,欧玉玲,吕春秋
(1.广西出入境检验检疫局检验检疫技术中心,广西 南宁 530021;2.桂林理工大学 化学与生物工程学院,广西 桂林 541004;3.广西中环能检测技术有限公司,广西 南宁 530031;4.贺州出入境检验检疫局,广西 贺州 542899)
碳点(Carbon dots,CDs),是近年来出现的一种新型荧光碳纳米粒子[1],具有sp2杂化碳的骨架结构,表面带有大量碳的含氧基团(羟基、羧基等)[2-3],且呈现出优异的荧光性能[4]。碳点的荧光量子产率比多数半导体量子点低,但具有优异的化学惰性、光稳定性、低毒性和良好的生物相容性,从而在光催化[5]、生物成像[6-7]、化学分析[8-9]等领域得到广泛应用。目前,碳点的合成方法主要有自下而上法(Bottom-up)[10-11]和自上而下法(Top-down)[12-13]。自下而上合成法包括高温热解法、微波合成法、水热法等。自上而下合成法包括电化学氧化法、激光消融法等。然而,已报道的方法虽能成功制备出性能良好的碳点,但大多需复杂的设备、苛刻的反应条件和昂贵的原料,不利于碳点的长期发展。
近年来,以自然界中随处可见的材料作为碳源合成碳点的方法引起了众多学者的高度关注。Tian等[14]以天然气燃烧的灰烬作为碳源,与5 mol·L-1的HNO3通过回流合成CDs,所合成的CDs粒径为(4.8±0.6) nm,最大激发、发射波长分别为310 nm和420 nm,但其所用的天然气燃烧灰烬不易获得。Lin等[15]以燃烧的废纸灰烬作为碳源,一步水热法合成了CDs,并建立了荧光“开关”用于有机磷农药的检测,所合成CDs在320 nm处出现明显吸收峰,且荧光发射光谱峰位置随激发波长的改变而变化,最大激发和发射光谱分别为320 nm和420 nm。
近年来农药的广泛使用,导致其在田间水体、土壤、食品中的残留问题日益突出。绿麦隆化学名为[N-(3-氯-4-甲基苯基)-N’,N’-二甲基脲],分子式为C10H13ClN2O,属接触性除草剂,用于农田中禾本科杂草和阔叶杂草的防除。绿麦隆是农田的重要污染之一,其在田间水体和土壤中的残留量过高会对农作物产生药害作用,还可对动物产生不同程度的毒害。
本文在前人研究的基础上,建立了水热法合成碳点的新方法。以常见的草木炭为碳源,通过与过氧化氢溶液水热反应,合成了高品质的水溶性CDs,所合成CDs的最大激发和发射波长分别为340 nm和450 nm。与文献[15]相比,本文合成的CDs无明显的紫外吸收,且荧光光谱不随激发波长的改变而改变。以得到的碳点作为荧光探针与绿麦隆相互作用,两者之间的电子转移使得碳点的荧光发生猝灭,且猝灭程度在一定范围内与绿麦隆浓度呈线性相关,据此建立了一种测定绿麦隆的新方法。
1 实验方法
1.1 仪器与试剂
RF-5301PC荧光光度计(日本岛津公司),TU-1901双光束紫外可见分光光度计(北京普析通用仪器有限责任公司),PHS-3C精密pH计(上海雷磁仪器厂),Avatar370傅立叶红外光谱仪(美国Nicola公司),FA1104电子分析天平(泰兴市电子仪器厂)。
草木炭:露天燃烧树枝(桂树枝、柏树枝、樟树枝、杂草、混合燃烧的草木碳),待其基本碳化后,通过研钵研磨成细碎粉末状,过100目筛,得到粒度较小的草木炭,密封保存于封口袋内。
过氧化氢溶液(陇西科学股份有限公司)、氨水(爱廉化试剂有限公司)、盐酸(爱廉化试剂有限公司)、罗丹明B(上海国药集团)、绿麦隆原药(纯度>97.0%,江苏快达农化有限公司)。所用试剂均为分析纯,实验用水为二次蒸馏水。
1.2 实验方法
1.2.1碳点的合成在20 mL聚四氟乙烯反应釜中,加入0.01~0.20 g自制粉末状草木炭和10 mL 1.0~5.0 mol·L-1的过氧化氢溶液,于120~200 ℃真空干燥箱中恒温加热2~12 h,冷却至室温后取出反应釜,然后过0.22 μm水系微孔滤头以除去未完全反应的草木炭及其他杂质,于2 000 Da透析袋中透析24 h以除去小颗粒与杂质,得到黄色透亮溶液。将得到的碳点溶液避光储存于4 ℃冰箱中,备用。
1.2.2碳点荧光猝灭法测定绿麦隆于一系列10 mL比色管中加入一定量的碳点和绿麦隆溶液,用水定容,25 ℃放置反应5 min后,于λex=340 nm,λem=450 nm处测定碳点的相应荧光强度(IF),仪器的激发和发射狭缝宽度均为5 nm。
2 结果与讨论
2.1 碳点合成条件的优化
2.1.1草木碳种类的选择考察了不同草木碳种类(桂树枝、柏树枝、樟树枝、杂草、混合燃烧的草木碳)对碳点荧光强度的影响。将草木碳研磨,过100目筛后,称取0.1 g按照“1.2.1”方法进行实验。结果表明,不同类型的草木灰对碳点的荧光强度无显著影响。文献[6]认为经燃烧或碳化的植物灰烬中含有大量的单质碳粒子,这些粒子经过热解、聚合、表面钝化等处理后形成了CDs,并发出荧光。
2.1.2草木炭用量的影响为考察草木炭用量对碳点发光的影响,分别称取0.01~0.20 g草木炭,在2.4 mol/L过氧化氢溶液中,于160 ℃下水热反应6 h制备碳点。结果表明,随着草木炭用量的增加,碳点的荧光强度先升高后降低。当草木炭用量为0.10 g时碳点的荧光强度最大。这可能是由于反应釜的空间一定(均为20 mL),随着草木炭用量的增多,单位空间内碳点的生成浓度增加。但继续增加草木炭的用量,反应釜内CDs的密度增大,达到饱和后过多的碳源不再生成CDs,反而加剧了分子间的碰撞,导致荧光发生猝灭。所以,本实验选择草木炭用量为0.10 g。
2.1.3过氧化氢浓度的影响称取0.10 g草木炭分别加入10 mL 1.0~5.0 mol·L-1的过氧化氢溶液中,于160 ℃下水热反应6 h制备碳点。结果表明,随着过氧化氢溶液浓度的升高,碳点的荧光强度先升高后降低,当过氧化氢浓度为2.4 mol·L-1时,CDs的荧光强度最强。因此,本实验最终选择加入2.4 mol·L-1过氧化氢。

图1 反应溶液pH值的影响Fig.1 Effect of pH value on reaction solution
2.1.4反应溶液pH值的影响实验对合成碳点反应溶液的pH值进行了探讨(图1),结果表明,在pH 2.0~5.0之间,荧光强度随pH值的增大而增强;pH>5.0时,荧光强度随pH值的增大而降低;pH=5.0时,荧光强度最大。且随着pH值升高,碳点的荧光光谱曲线出现不同程度的蓝移,曲线的峰宽变宽,说明碳点溶液中的杂质增多。这可能是由于氧化剂的主要成分被分解所致。过氧化氢溶液的主要成分为H2O2,pH值升高,过氧化氢分解的速度加快(尤其在70 ℃下)[16],其反应式为:2H2O2→ 2H2O + O2。因此,反应溶液的pH值对过氧化氢溶液的稳定性存在影响,降低pH值,可增加过氧化氢溶液的稳定性。为保证反应的顺利进行和前预处理的简易化,本实验选择反应溶液的pH值为5.0,此条件下反应后碳点溶液的pH值约为7.0。
2.1.5水热温度的影响称取0.10 g草木炭加入10 mL 2.4 mol·L-1的过氧化氢溶液中,分别于120~200 ℃下水热反应6 h制备得到碳点溶液,以考察水热温度对碳点荧光强度的影响。结果表明,当温度从120 ℃升至160 ℃时,碳点的荧光强度不断增强。继续升高温度,其荧光强度下降,发射峰峰位蓝移且出现变形。这可能是因为随着温度的上升,反应越来越充分,碳点的浓度增加,荧光强度随之增强。但温度继续上升后,生成的碳点颗粒过分加热,表面结构被破坏,从而导致荧光发射量减少[17]。此外,温度对碳点的粒径也存在影响,所以碳点的发射峰出现蓝移,峰宽有所增加。因此,本实验最终选择160 ℃为CDs合成的反应温度。
2.1.6水热时间的影响称取0.10 g草木炭加入10 mL 2.4 mol·L-1的过氧化氢溶液中,于160 ℃下分别水热2~12 h制备得到碳点溶液,考察了水热时间的影响。结果表明,随着水热反应时间的增加,碳点的荧光强度先升高后下降。反应6 h时,碳点的荧光强度最强,继续延长反应时间,荧光强度反而下降。这可能是因为反应时间过长,碳点的表面结构被破坏,从而导致荧光发射量减少,荧光强度下降。故选择水热时间为6 h。
2.2 碳点的表征
2.2.1CDs的紫外吸收光谱与荧光发射光谱图2为碳点的紫外-可见吸收光谱和荧光发射光谱。以草木炭为碳源合成的碳点在可见光区内无显著的吸收峰(图2A,曲线a)。随着碳点的发射光谱从310 nm增至380 nm,碳点的荧光强度先升高后降低,碳点的最大激发波长340 nm和350 nm处的发射峰几乎重叠,最大发射波长约为450 nm,随着激发波长的改变,发射波长未出现明显的红移或蓝移(图2B)。

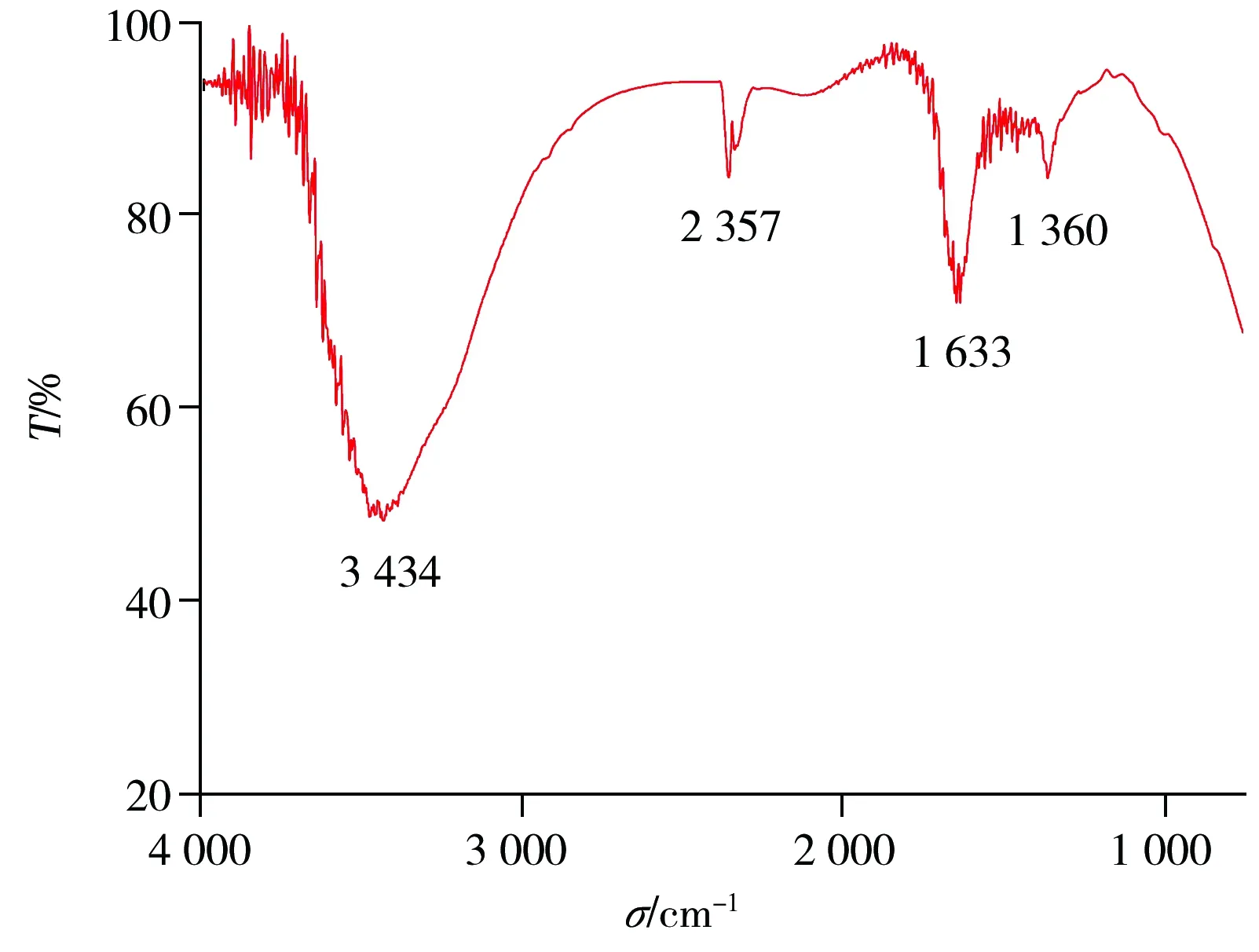
图3 碳点的傅立叶变换红外光谱Fig.3 Fourier transform infrared(FTIR) spectra of carbon dots
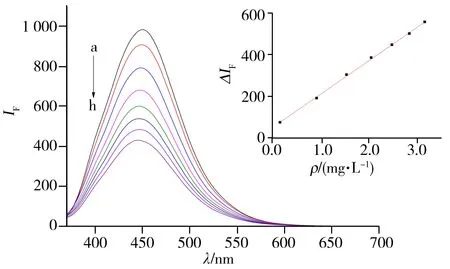
图4 绿麦隆对碳点的荧光猝灭曲线Fig.4 Fluorescence spectra of chlorotoluron quenched by carbon dotsinsert:standard work line;ρchlorotoluron(a-h):0,0.15,0.91,1.53,2.04,2.47,2.83,3.15 mg·L-1

图5 CDs与绿麦隆的相互作用机理Fig.5 Interaction between CDs and chlorotoluron

2.3 绿麦隆的碳点荧光猝灭法测定
按照“1.2.2”方法,考察了绿麦隆对碳点的猝灭作用。如图4所示,随着绿麦隆加入量的增加,碳点的荧光强度降低。由图4可知,绿麦隆的质量浓度(ρ)在0.15~3.15 mg·L-1范围内与碳点在450 nm处的荧光猝灭程度(ΔIF)呈线性关系,回归方程为ΔIF=160.2ρ+51.74,r2=0.999 3,计算得到检出限(S/N=3)为0.05 mg·L-1,据此建立了一种测定绿麦隆的新方法。
2.4 绿麦隆对碳点的荧光猝灭机理

2.5 干扰实验

2.6 实际样品的测定
取不同农田的水样,静置过夜后取上清液过滤以除去固体杂物,滤液置于洁净的容器中,待测。实验中水样的取样量均为500 μL。若需进行快速分析,可将水样离心后取上清液按“1.2.2”方法进行测定,同时进行加标回收实验,结果如表1所示。由表1可知,样品测定结果的相对标准偏差(RSD)不大于4.9%,加标回收率为93.3%~112%,说明所建立的方法具有较高的准确度和精密度,可应用于实际农田水样品中绿麦隆含量的分析。

表1 样品测定结果Table 1 Measurement result of the sample
*no detected
3 结 论
本文建立了一种以草木炭为碳源的新型碳点合成方法。实验显示,0.10 g草木炭在2.4 mol/L过氧化氢溶液中,于160 ℃下水热反应6 h得到的碳点溶液荧光性能最好。红外光谱表明,碳点的表面分布着大量—COOH和—OH基团,故碳点在水性体系中表现出良好的稳定性和亲水性。由于绿麦隆能够有效猝灭碳点的荧光强度,因此利用所合成的荧光碳点建立了绿麦隆测定的新方法。该方法测定绿麦隆的线性范围为0.15~3.15 mg·L-1,检出限为0.05 mg·L-1,用于实际样品分析,其加标回收率为93.3%~112%,RSD不大于4.9%。该方法具有操作简单、线性范围宽、选择性好等特点,可应用于实际农田水样品中绿麦隆的检测,并为其他样品中绿麦隆的检测提供参考。
猜你喜欢
潍坊学院学报(2021年2期)2021-07-22
北方人(2019年24期)2019-12-19
少年漫画(艺术创想)(2019年6期)2019-10-12
分析化学(2017年12期)2017-12-25
分析化学(2017年12期)2017-12-25
科学大众·小诺贝尔(2016年6期)2016-08-17
无机化学学报(2014年12期)2014-02-28
无机化学学报(2014年8期)2014-02-28
无机化学学报(2014年4期)2014-02-28
东北师大学报(自然科学版)(2014年1期)2014-02-27
