
地球化学样品中钒检测方法的对比
2018-10-09 08:39:32王文杰刘安安
分析测试技术与仪器 2018年3期
王文杰,刘安安
(甘肃省有色金属地质勘查局 张掖矿产勘查院,甘肃 张掖 734000)
在自然界中,钒很难以单一体存在,主要与其他矿物形成共生矿或复合矿. 目前发现的含钒矿物有70多种,但主要的矿物有以下3种:钒钛磁铁矿、钾钒铀矿和石油伴生矿,现在已探明的钒资源储量的98%赋存于钒钛磁铁矿中,V2O5含量可达1.8%. 工业上广泛用于催化剂和制造优质合金刚.
钒是重要的地球化学指示元素,是全球地球化学填图、多目标生态地球化学调查等项目的必测元素,在检测过程中,钒和铬检测结果的准确性对于生态环境评价、矿产资源调查和人体健康及环境等均具有非常重要的意义[1]. 地球化学样品中钒的测定方法主要有原子吸收光谱法[2]、等离子体发射光谱法(ICP-AES)[3]、电感耦合等离子体质谱法(ICP-MS)[4-6]和X射线荧光光谱法[7-10]. 本文通过对4种方法的试验对比,进而做出对地球化学样品中钒的检测方法的讨论.
1 试验部分
1.1 原子吸收光谱法
1.1.1 仪器及分析条件
分析天平(梅特勒-托利多):感量0.000 1 g , 型号: AB54-S ;原子吸收光谱仪(美国PerkinElmer): AA400 ,工作参数如表1所列.
1.1.2 标准溶液和试剂
钒标准储备溶液:ρ(V)=1 000 μg/mL;钒标准工作溶液:20 mg/L,使用时由标准储备液逐级稀释制备.

表1 原子吸收光谱仪工作参数Table 1 Operating parameters of atomic absorption spectrometer
硝酸、盐酸、高氯酸、氢氟酸、磷酸均为优级纯,试验溶液用蒸馏水配制.
1.1.3 标准曲线的绘制
在6支25mL比色管中,分别吸取0、1.0、2.0、3.0、4.0、5.0 mL,质量浓度为20 mg/L的钒标准工作液,用质量分数为2%的磷酸溶液定容至刻度. 分别测定吸光度后随浓度作图,回归的曲线方程为A=0.010C-0.001,相关系数r(V)为0.999 2.
1.1.4 样品处理
准确称取0.500 0 g国家一级标准物质于聚四氟乙烯坩埚中,用少许水润湿,先分别加入10 mL HNO3和10 mL HF放置在电热板上低温加热至近干后,再分别补加5 mL HNO3、5 mL HF和2 mL高氯酸,继续加热至近干,加入5 mL HCL将盐分溶解后转移至25 mL比色管中,用质量分数为2%的磷酸定容至刻度,充分摇匀静止后取上层清液和标准系列溶液一起测定吸光度.
1.1.5 方法检出限
取20份样品空白溶液用本法测定,采用GB/T 27415-2013[11],求得V的方法检出限(LD)为0.8 μg/g.
1.1.6 方法精密度
选定5个国家一级标准物质,按照此检测方法测定12次V的含量,计算其相对标准偏差(RSD),结果如表2所列.
由表2结果可见,通过原子吸收光谱法测定地球化学样品中V的相对标准偏差为2.59%~3.37%,其相对标准偏差RSD均在10%以下,符合质量规范要求.
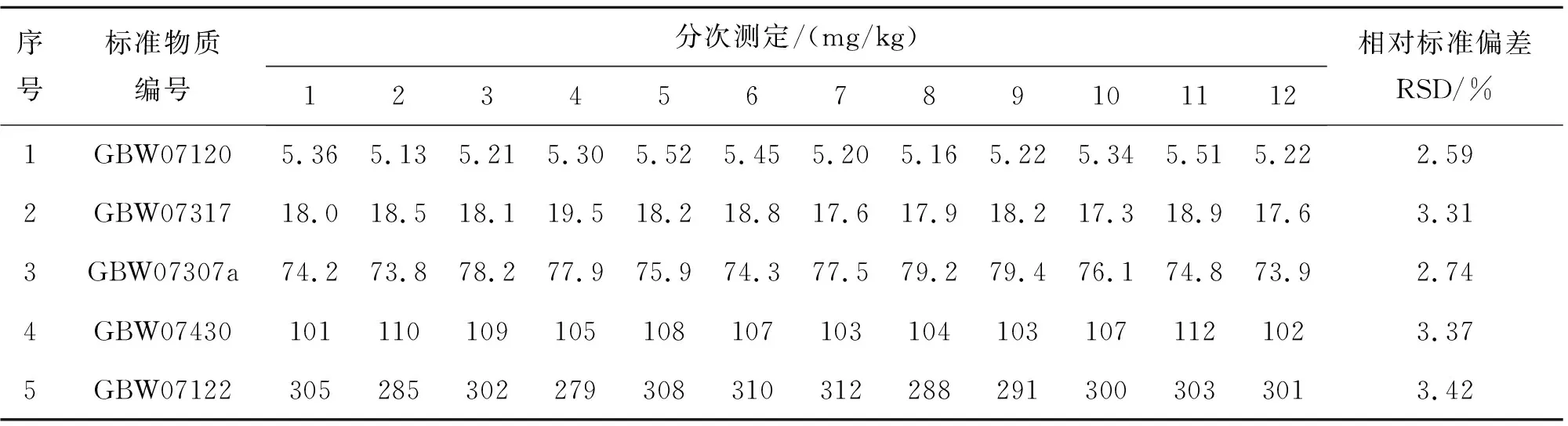
表2 方法精密度Table 2 Precision of method
1.1.7 方法准确度
选定5个国家一级标准物质,按照此检测方法测定V的含量,计算其相对偏差(RE),结果如表3所列.

表3 方法准确度Table 3 Accuracy of methods
表3结果表明,通过原子吸收光谱法测定地球化学样品中的V,测定值与标准值相吻合,证明该方法可靠.
1.1.8 方法加标回收率
按照此检测方法,进行标准样品加入法回收试验. 使用国家标准物质GBW07120、GBW07317、GBW07307a、GBW07430、GBW07122验证,样品回收率在96.5%~103.5%之间,结果如表4所列.
1.2 等离子体发射光谱法
1.2.1 仪器及分析条件
分析天平(梅特勒-托利多):感量0.000 1 g,型号:AB54-S;电感耦合等离子体光谱仪(美国热电公司):ICP-6300,工作参数如表5所列.
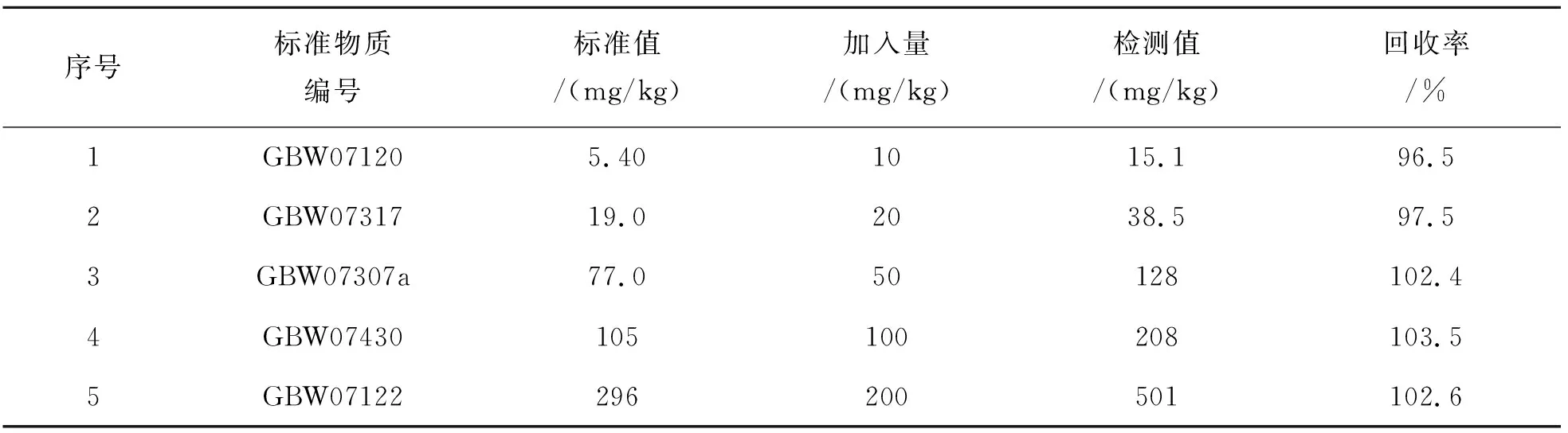
表4 加标回收率Table 4 Spiked recovery rate

表5 光谱仪工作参数Table 5 Operating parameters of spectrometer
1.2.2 样品处理
准确称取0.200 0 g国家一级标准物质于50 mL聚四氟乙烯坩埚中[12],加几滴水润湿. 加入10 mL HNO3、10 mL HF、5 mL HCL和2 mL HCLO4,置于控温电热板上,加坩埚盖,放置过夜. 次日,升温至110 ℃,保持1.5~2 h. 揭去盖子,升温至240 ℃,直至高氯酸白烟冒尽,加2 mL 质量分数为50% HCL,趁热浸取,冷却. 移入具有刻度的25 mL带塞塑料管中,用水稀释至刻度,摇匀. 待ICP-AES测定.
1.2.3 方法检出限
取20份样品空白溶液用本方法测定,采用GB/T 27415-2013[12],求得V的方法检出限(LD)为0.3 μg/g.
1.2.4 方法精密度
选定5个国家一级标准物质,按照此检测方法测定12次V的含量,计算其相对标准偏差(RSD),结果如表6所列.
表6结果表明,通过电感耦合等离子体发射光谱法测定地球化学样品中V的相对标准偏差为1.14%~4.04%,其相对标准偏差RSD均在10%以下,符合质量规范要求.
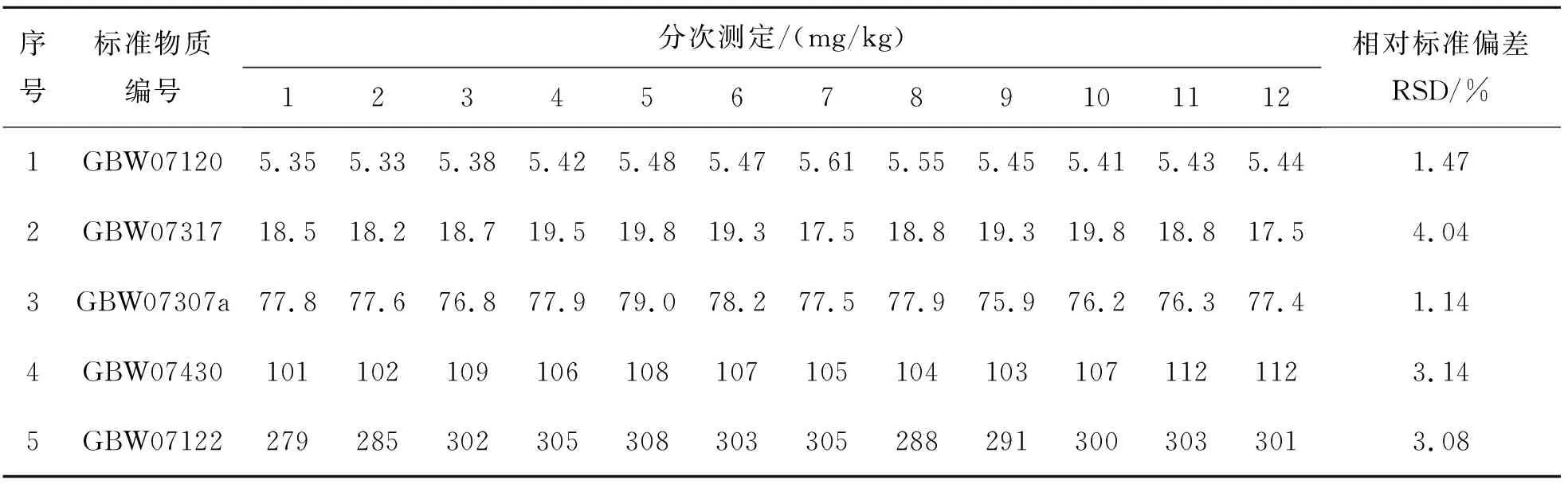
表6 方法精密度Table 6 Precision of method
1.2.5 方法准确度
选定5个国家一级标准物质,按照此检测方法测定V的含量,计算其相对偏差(RE),结果如表7所列.
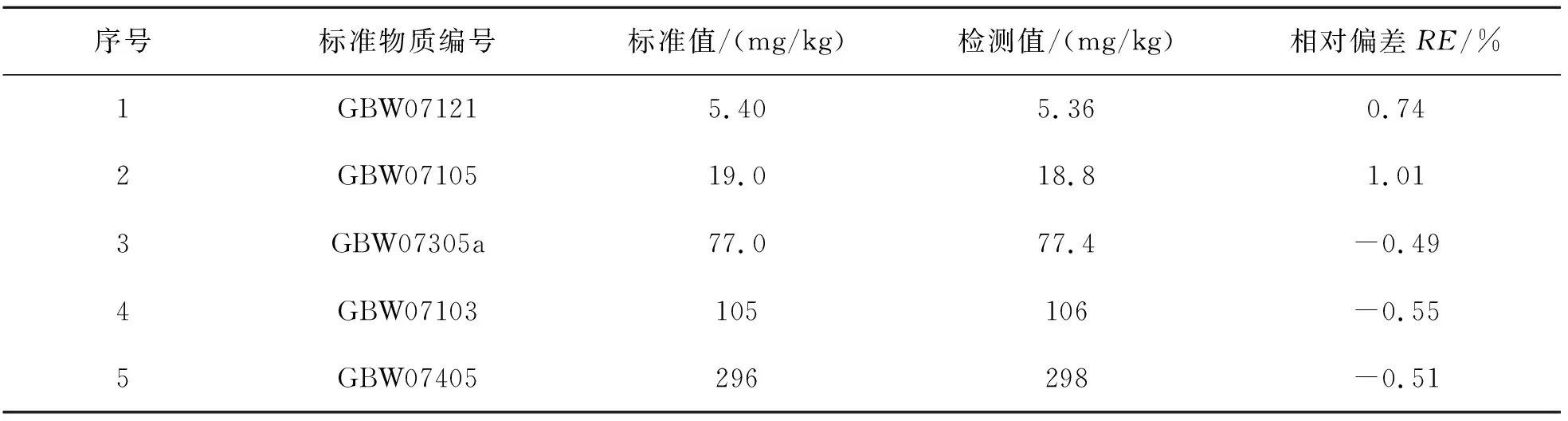
表7 方法准确度Table 7 Accuracy of methods
表7结果表明,通过电感耦合等离子体发射光谱法测定地球化学样品中的V,测定值与标准值相吻合,证明该方法可靠.
1.2.6 方法加标回收率
按照此检测方法,进行标准样品加入法做回收试验. 使用国家标准物质GBW07120、GBW07317、GBW07307a、GBW07430、GBW07122验证,样品回收率在95.5%~105.1%之间,结果如表8所列.
1.3 电感耦合等离子体质谱法(ICP-MS)
1.3.1 仪器及分析条件
分析天平(梅特勒-托利多):感量0.000 1 g,型号:AB54-S;电感耦合等离子体质谱仪(美国热电公司):iCAP-Qa, 工作参数如表9所列.
1.3.2 样品处理
准确称取0.200 0 g国家一级标准物质于50 mL聚四氟乙烯坩埚中,加几滴水润湿. 加入10 mL HNO3、10 mL HF和2 mL HCLO4,放置在控温电热板上,升温至200 ℃,蒸发至高氯酸冒烟约10 min,取下冷却;再依次加入5 mL HNO3、5 mL HF和1 mL HCLO4于电热板上加热15 min后关闭电源,放置过夜后,再次加热至高氯酸烟冒尽[13]. 趁热加入5 ml王水(1+1),在电热板上加热至溶液体积剩余2~3 mL,用水冲洗杯壁,微热5~10 min至溶液清亮,取下冷却. 将溶液转移至20 mL有刻度的聚乙烯试管中,用去离子水定容至刻度,摇匀,澄清. 移去清液1.00 mL置于聚乙烯试管中,用质量分数为3% HNO3稀释至10.0 mol/L,摇匀,待测.
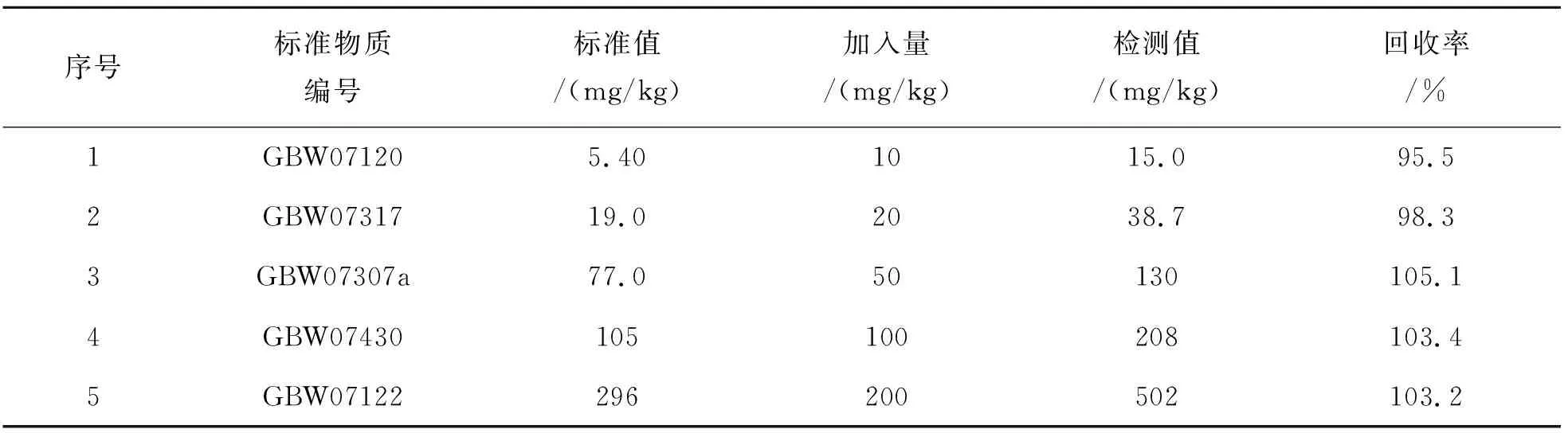
表8 加标回收率Table 8 Spiked recovery rate
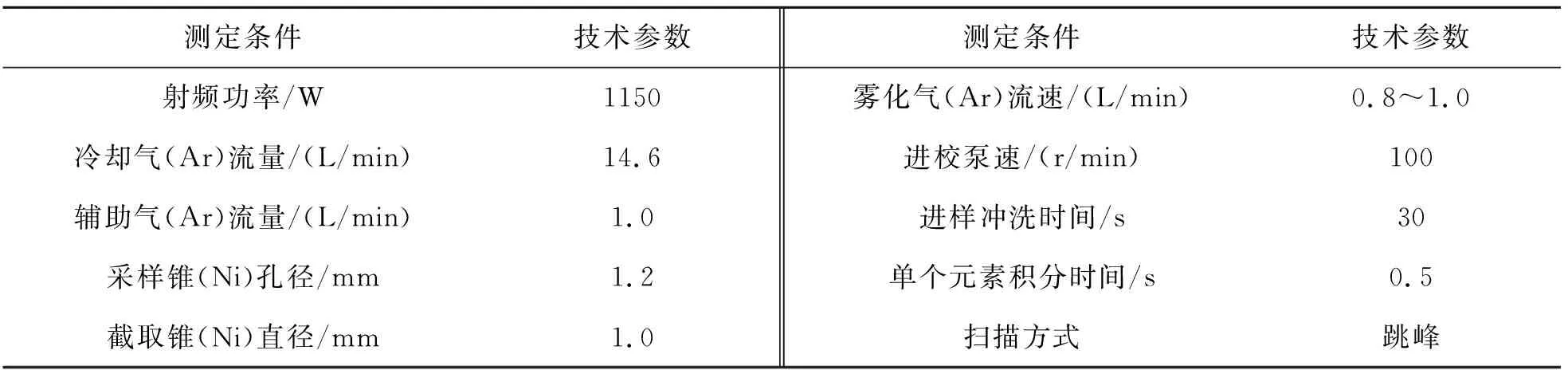
表9 等离子体质仪工作条件Table 9 Working conditions of plasma mass spectrometer
1.3.3 方法检出限
取20份样品空白溶液用本方法测定,采用GB/T 27415-2013,求得V的方法检出限(LD)为0.2 μg/g.
1.3.4 方法精密度
选定5个国家一级标准物质,按照此检测方法测定12次V的含量,计算其相对标准偏差(RSD),结果如表10所列.
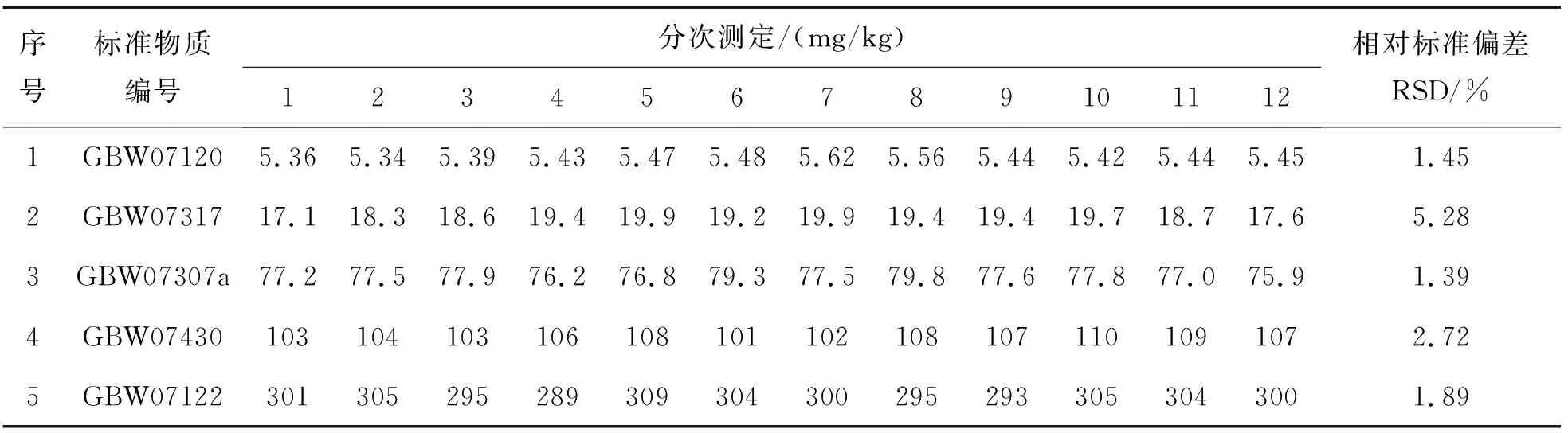
表10 方法精密度Table 10 Precision of method
表10结果表明,通过电感耦合等离子体质谱法测定地球化学样品中V的相对标准偏差为1.45%~5.28%,其相对标准偏差RSD均在10%以下,符合质量规范要求.
1.3.5 方法准确度
选定5个国家一级标准物质,按照此检测方法测定V的含量,计算其相对偏差(RE),结果如表11所列.
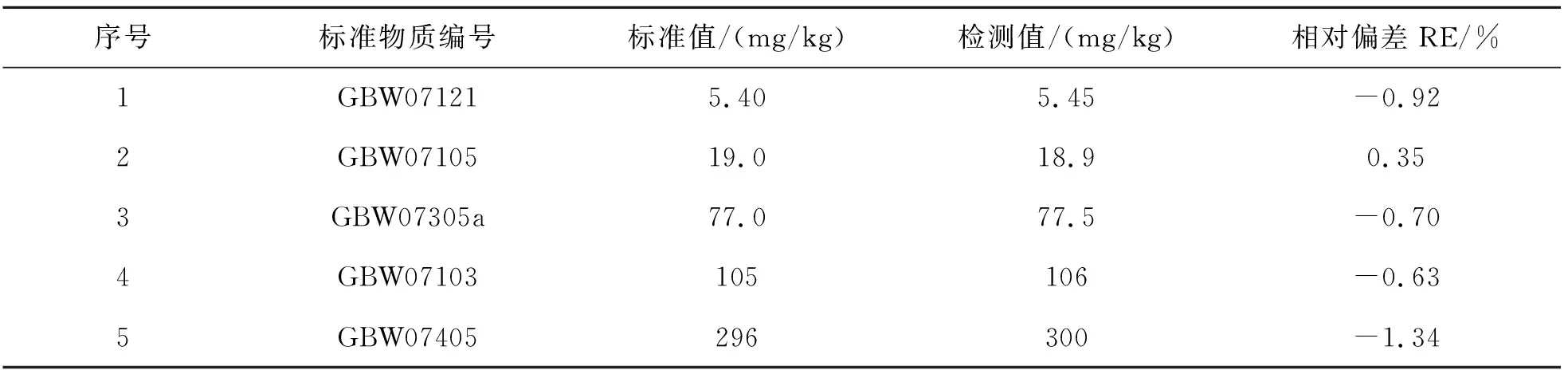
表11 方法准确度Table 11 Accuracy of methods
表11结果表明,通过电感耦合等离子体质谱法测定地球化学样品中的V,测定值与标准值相吻合,证明该方法可靠.
1.3.6 方法加标回收率
按照此检测方法,进行标准样品加入法做回收试验. 采用国家标准物质GBW07120、GBW07317、GBW07307a、GBW07430、GBW07122验证,样品回收率在96.8%~106.8%之间,结果如表12所列.
1.4 X射线荧光光谱法
1.4.1 仪器及分析条件
X荧光光谱仪(荷兰帕纳科):Axios-mAX型,以 Rh靶X-射线管为激发源 ,SuperQ软件 ,压片粉末:聚乙烯粉,样品杯:钢杯 37 mm. 具体测试条件如表13所列.

表12 加标回收率Table 12 Spiked recovery rate

表13 XRF中V元素测试条件Table 13 Test conditions for V elements in XRF
1.4.2 样品处理
样品倒入模具内,用低压聚乙烯粉镶边衬底,置于压力机上压制成型. 试料片的外径为40 mm,内径为31 mm,压制完成的试料片在非测量面贴上标签或用记号笔编写样号,放入干燥器内保存,以防吸潮和污染.
1.4.3 方法检出限
考虑到含量计数率、背景计数,样品精密度、准确度,根据GB/T 27415-2013定量限评估要求,确定利用粉末压片-XRF测定V的测定下限为4.8 μg/g.
1.4.4 方法精密度
选定5个国家一级标准物质,按照此检测方法测定12次V的含量,计算其相对标准偏差(RSD),结果如表14所列.
表14结果表明,通过X射线荧光光谱法测定地球化学样品中V的相对标准偏差为1.41%~6.60%,其相对标准偏差RSD均在10%以下,符合质量规范要求.
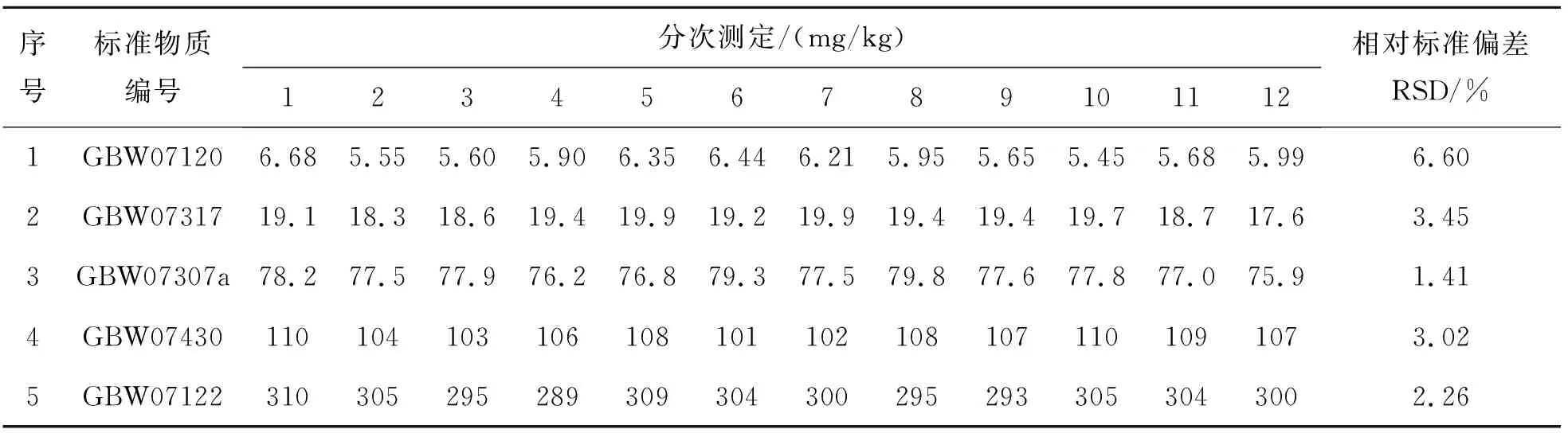
表14 方法精密度Table 14 Precision of methods
1.4.5 方法准确度
选定5个国家一级标准物质,按照本法测定V的含量,计算其相对偏差(RE),结果如表15所列.
表15结果表明,通过X射线荧光光谱法测定地球化学样品中的V,测定值与标准值相吻合,证明该方法可靠.
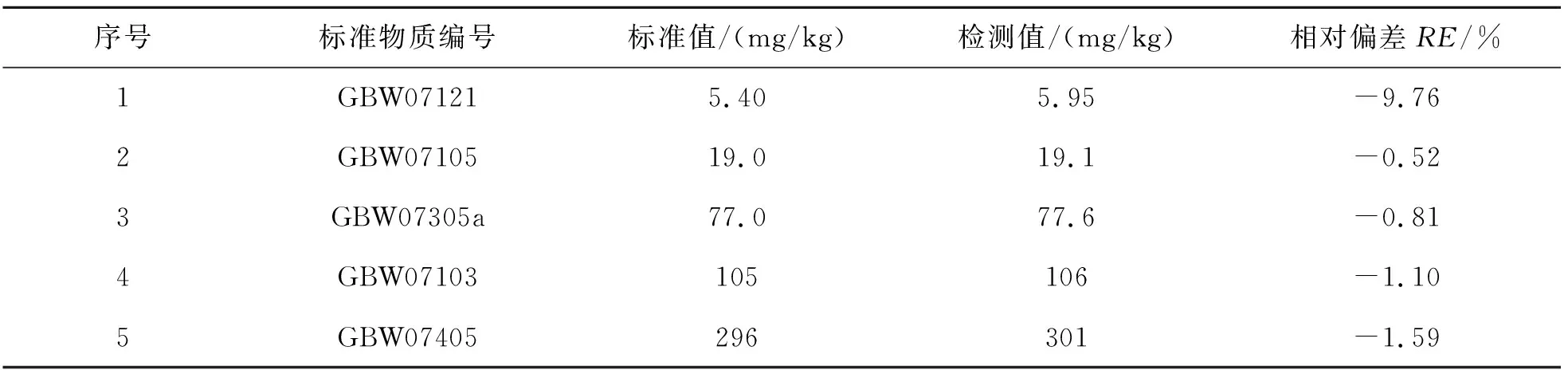
表15 方法准确度Table 15 Accuracy of methods
2 结论
本文通过对4种方法试验数据的对比,表明其测定结果均满足DD2005-03[14]对化探样品中V的试验要求. 其中,原子吸收法检出限较高,分析速度慢. 电感耦合等离子体质谱法精密度高,检出限低,线性范围广,但对质谱仪分辨率要求较高,而且存在质谱干扰,CL16O对51V存在严重干扰,无法直接测定,需用碰撞/反应池技术消除原子离子干扰. X射线荧光光谱法检出限较高,低含量样品测试结果准确度差,适合含量较高的样品. 等离子体发射光谱法检出限低,线性范围宽,精密度好,谱线干扰较小,分析速度快,更适合地球化学样品中钒的批量生产.
猜你喜欢
科学技术创新(2021年19期)2021-07-16 10:07:18
World Journal of Clinical Cases(2020年17期)2020-09-18 08:03:24
检验医学与临床(2020年1期)2020-01-10 04:44:22
建筑科技(2018年6期)2018-08-30 03:40:54
中国交通信息化(2016年5期)2016-06-06 03:51:43
科技与创新(2016年10期)2016-05-28 03:17:18
应用海洋学学报(2015年2期)2015-11-22 07:36:40
现代检验医学杂志(2015年6期)2015-02-06 01:44:25
测绘通报(2014年3期)2014-08-16 03:15:52
天津冶金(2014年4期)2014-02-28 16:52:58
