
一种新型的构建CRISPR/Cas9 KnockOut载体的方法
2018-10-08 11:53陈恒玲黄玉连林显光
中南民族大学学报(自然科学版) 2018年3期
陈恒玲,许 杰,黄玉连,林显光
(中南民族大学 生物医学工程学院,武汉430074)
随着测序技术以及大数据信息科学的发展,越来越多疾病的致病基因突变已经被发现.同时如今的技术已经可以预测一些疾病的发生可能与基因组上哪些SNP突变相关.然而,需要验证预测的结果,或是对已经发生的基因突变进行治疗,就必须依靠基因编辑技术[1].
CRISPR系统最早是在Streptococcuspyogenes细菌的获得性免疫系统中发现的[2].在临近CRISPR 的区域包含一组保守的蛋白质编码基因,称为CRISPR-associated基因,即Cas基因,其编码的蛋白质包含解旋酶、聚合酶、核酸酶等[3].
CRISPR编辑基因组被称为第三代基因编辑技术,即RNA指导的Cas9核酸酶对目标基因进行编辑的技术[4].CRISPR编辑基因组只需要在细胞中导入合成的sgRNA表达载体,即可配合同时表达的Cas9核酸酶,对任何物种的基因组进行高效率的定向编辑(如图1所示).因此CRISPR具有高度特异性,不需要引入外源DNA即可在任何细胞中剔除、插入、甚至是突变想要的多个基因[4].相较于传统的ZFN系统与TALENs而言,CRISPR有着更多的优点:
没有物种限制,可在动物、植物、细菌等生物中进行基因打靶.载体构建简单快速.靶位点设计容易,PAM 位点(NGG)于128 bp随机DNA 序列中就会出现一次,并且正反两链皆可设计.Cas9 辨识DNA 序列效率高,有效降低脱靶效应,增加打靶准确性.基因打靶成功率较ZFN 或TALEN 高,还可同时放入两个以上的sgRNA 来进行多个基因同时剔除的目的[5].双合一或三合一载体简化细胞转染的难度.搭配多种报导基因,如荧光蛋白或抗生素筛选基因,可以直接作用于DNA 上,让基因沉默更加简单,将可渐渐取代RNA(RNAi)等基因干扰技术[6].
近几年来,利用 CRISPR技术构建小鼠模型以及修复小鼠疾病模型在医学研究各大领域均有非常迅速的发展[7].例如:利用CRISPR/Cas9 基因编辑系统将各种致癌突变导入到成年小鼠的肝脏中,构建癌症模型,使科学家能够更快速地筛查这些突变以及这些不同突变相互作用产生癌症的机制,并且还能研究一些药物对肿瘤所产生的潜在效应[8].
本项目拟借用LentiCRISPRv2的载体,并用一种新型的KO载体构建方式,获得Spata49基因的KnockOut载体[1, 9].LentiCRISPRv2 的载体是张锋实验室发明的一种整合了慢病毒包装元件的新型CRISPR/Cas9载体,其重组载体可以直接通过三代慢病毒包装系统进行包装(如图2所示).其标准的载体构建方式为将合成的oligo进行磷酸化修饰,然后通过T4连接酶进行连接[10].而本文以小鼠的Spata49(Spermatogenesis associated 49,即4933417A18RiK,GeneID 66761)为例,说明我们实验室发明的一种新型的基于退火方式的载体构建方式.
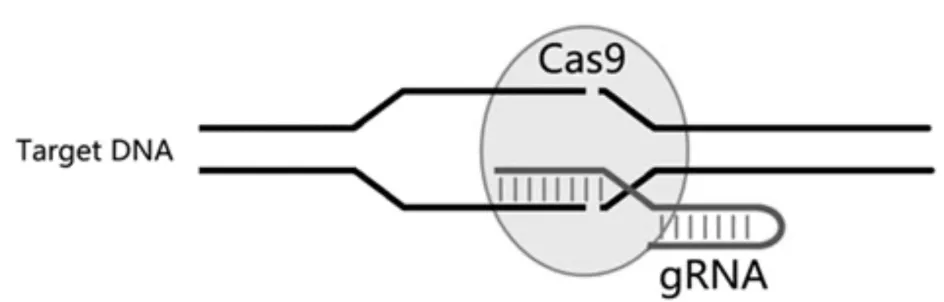
图1 RNA指导的CRISPR/Cas9基因剪切系统Fig.1 CRISPR/Cas9 KnockOut system guided by RNA

图2 LentiCRISPR V2质粒图谱Fig.2 Plasmid profiles of LentiCRISPR V2
1 材料与方法
1.1 实验材料
Spata49(单链oligo1和oligo2,武汉tsingke公司合成)、Annealing Buffer(碧云天)、质粒提取试剂盒(Omega)、BsmBⅠ酶(Thermo)、DNA凝胶回收试剂盒(Omega)、连接酶(NEB)、CaCl2感受态细胞(Stbl3菌种实验室制备).
1.2 实验方法
1.2.1 基因敲除靶点和寡核苷酸设计
1)基因靶点选择.CRISPR基因敲除系统靶点由25个碱基组成[8].靶点前面为起始转录信号G,在设计基因敲除的时候,在基因起始密码子附近与下游找相应序列作为靶点,如果没有找到可以选择起始信号,构建载体的时候再加一个G作为启动信号[8].
2)插入寡核苷酸序列设计.本次课题引物设计选择的是 Zhang Lab 的在线设计软件:从http://crispr.mit.edu/.在线得到sgRNA,由sgRNA模板序列设计互补的DNA oligo,本次课题选择两条引物序列为5′ CACCGTGTGATTCAAGACGTCAGGC 3′,3′CACACTAAGTTCTGCAGTCCGCAAA 5′(即5-AAACGCCTGACGTCTTGAATCACAC-3).如图3所示,黑体表示靶基因序列,下划线表示要酶切后的载体相补的部分,即接头.

图3 Oligo序列示意图Fig.3 Oligo sequence schematic diagram
1.2.2 酶切
根据提取LentiCRISPRv2质粒的浓度和取样跑胶结果来决定酶切所需要的最佳质粒,表1为本次课题酶切的体系,条件为在37 ℃环境中切3 h.这个条件是在前期实验中得到的比较合适BsmBⅠ酶的酶切条件.
注:酶切以后回收大约11000 bp的条带.

表1 酶切体系Tab.1 Enzymedigest system
1.2.3 退火
将引物合成公司送来的Spata49(oligo1、oligo2)稀释为100 μmol/L的浓度.进行退火实验,其退火体系如表2所示.

表2 退火体系Tab.2 Annealing system
注:1)95 ℃降至室温的时候要严格控制温度的变化, 具体是每分钟1 ℃降温;2)退火后的引物需要和水进行1:200的稀释
1.2.4 连接
将已经退火好的引物和回收的DNA进行连接实验,其条件为4 ℃过夜连接或者16 ℃连接1 h.其连接体系如表3所示.

表3 连接体系Tab.3 Ligase reaction system
1.2.5 转化
1)取连接好的产物3 μL加于100 μL的感受态细胞内,均匀混合后,在冰上静置30 min.
2)准备42 ℃水浴锅、37 ℃预热800 μL的LB培养液,将冰上的混合物放在42 ℃水浴锅90秒,立即拿出放在冰上3 min,然后在超净台中将LB培养液加入混合液.
3)将混合液放入37 ℃ 低于100 r/min恒温摇床中45 min.
4)将摇床后的混合液5000 g离心5 min,去大部分上清液,留下大概100 μL的培养基悬浮细胞并将细胞吹散,最后将转化的细胞均匀涂在LB平板上面,10 min后,待液体完全吸收后将平板倒置,在37 ℃培养箱中培养12 h.
1.2.6 取样送公司测序
本实验用的引物为LKO.1.5,序列为5′GACTATCATATGCTTACCGT 3′.
2 结果及讨论
2.1 质粒提取和质粒酶切的结果
LentiCRISPRv2提取浓度为295 ng/μL, A260:A280为1.960,符合下一步的实验要求.酶切后跑胶的结果如图4所示,主带成线性化,且有一条2 kb的带出现,说明酶切成功.
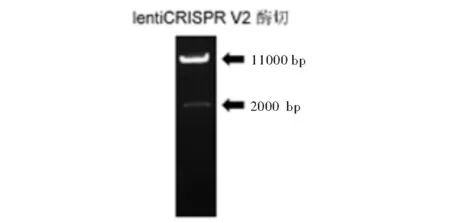
图4 质粒酶切跑胶Fig.4 Plasmids enzyme digestion gel
2.2 转化与测序鉴定
根据培养好的LB平板挑取菌斑,送样公司测序后,根据公司反馈的结果和原序列对比发现载体构建成功,其碱基峰值图如图5所示,框中所示为sgRNA插入序列,即剪辑靶位点序列.
3 结语
本文提出了一种基于oligo退火的CRISPR/Cas9的载体构建方式,该方式不仅可以应用于LentiCRISPRv2的载体,其类似的载体都可以采用.在标准操作中,载体用碱性磷酸酶去磷酸化处理,oligo在T4 PNK作用下进行磷酸化修饰,同时进行连接反应.这种方式的优点是载体进行去磷酸化处理,避免载体自连带来的假阳性.但是具体到CRISPR/Cas9的载体,BsmBⅠ酶切产生的切头并不互补,不会自连,所以载体不进行去磷酸化处理不会对后续实验产生影响.而假阳性的产生最重要的原因是载体酶切不完全.结果表明:我们的方法具有以下优势.
1) 因为直接合成oligo,没有进行磷酸化的修饰,也不需要T4 PNK处理,其价格低廉.
2) 用退火的方式代替连接反应,其操作较为方便.
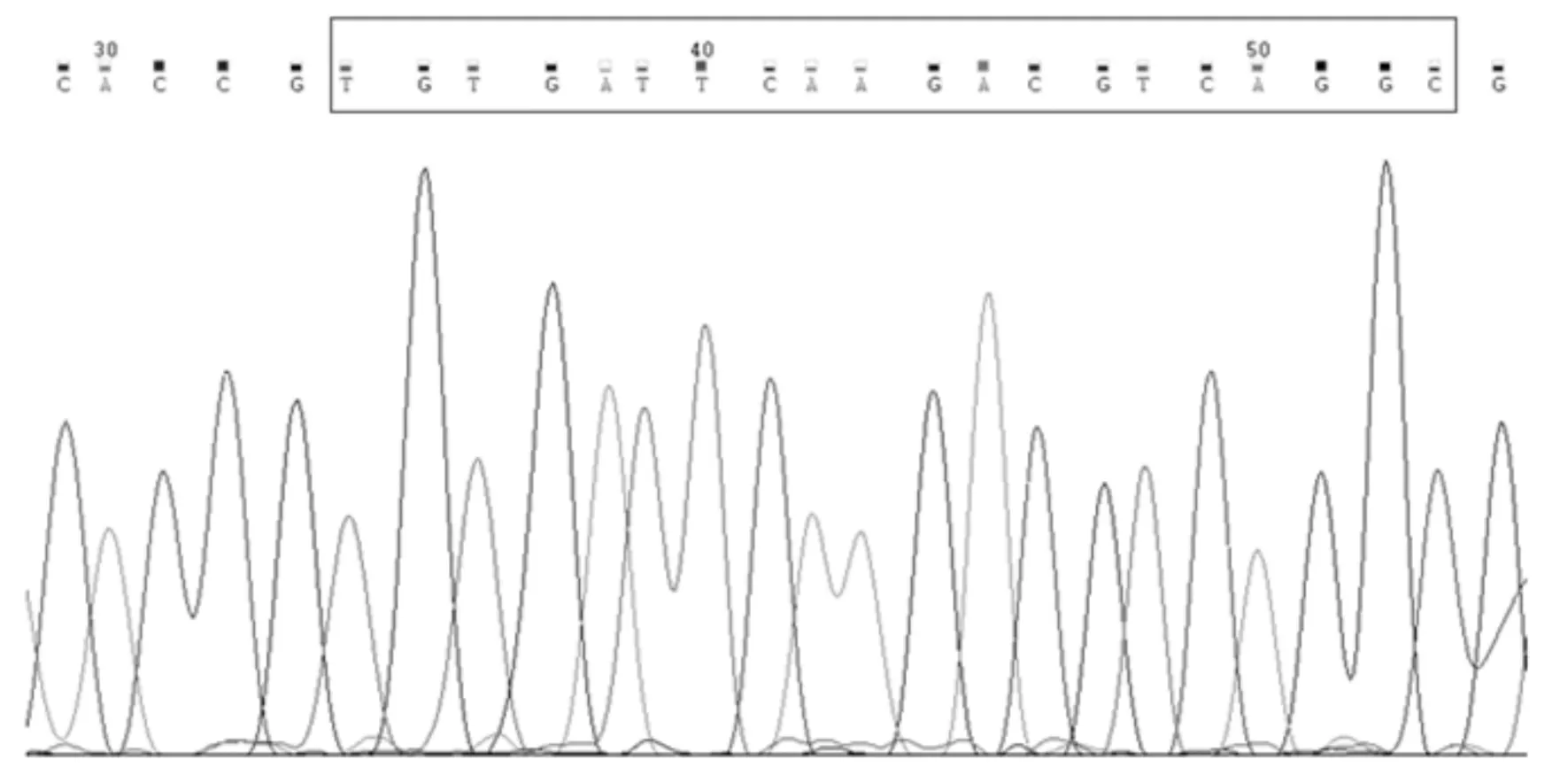
图5 Spata49的KO载体碱基峰值图Fig.5 Spata49 KO vector sequencing map
猜你喜欢
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
成都医学院学报(2022年4期)2022-08-19
波谱学杂志(2022年1期)2022-03-15
昆明医科大学学报(2022年1期)2022-02-28
中老年保健(2021年3期)2021-12-03
江西农业学报(2021年4期)2021-04-20
中国生殖健康(2020年7期)2020-12-10
三农资讯半月报(2020年11期)2020-06-21
分析化学(2017年12期)2017-12-25
安徽医科大学学报(2015年9期)2015-12-16
