
常染色体隐性遗传性皮肤松弛症的相关基因研究进展
2018-09-28 03:49:00石武娟薛珊珊
天津医药 2018年9期
石武娟,薛珊珊
皮肤松弛症(cutis laxa,CL)是指因细胞外基质中的弹力蛋白合成受阻或结构异常引起的一种症候群,是一种罕见的结缔组织病[1]。该病的典型特征为非衰老所致的皮肤弹性下降、松弛及下垂,其中以面部皮肤受累最为多见,可伴有眼睑、脸颊及颈部皮肤的松弛和下垂。同时,患者还可伴有其他富含弹性纤维的组织器官的功能障碍[2],如血管、心脏、肺等。因为皮肤松弛症的遗传方式和临床表现具有多样性,所以给患者及时准确的诊治带来一定的困难与挑战。本文旨在综述近年来常染色体隐性遗传性皮肤松弛症发病机制的研究进展,从而为该病的诊治及预防提供新的思路。
1 皮肤松弛症的分类
按遗传特征,可将皮肤松弛症分为遗传性和获得性两大类。获得性皮肤松弛症是指某些因素如感染或药物等作用使弹性蛋白结构和(或)数量发生不同程度的病理改变,最终导致皮肤弹性减退以致松弛[3-5]。获得性皮肤松弛症通常表现为局部病灶,且在发病前无皮肤损害的表现,但亦可出现全身受累[5]。与获得性皮肤松弛症相比,遗传性皮肤松弛症通常发病早,在出生时或婴儿期即可发病。遗传性皮肤松弛症的发病率约为1/40万~1/20万[1],主要分为常染色体显性遗传、常染色体隐性遗传及X连锁隐性遗传三种遗传方式。
2 皮肤维持弹性的生物学基础
皮肤的细胞外基质由多种成分共同构成,包括胶原、蛋白多糖、层粘连蛋白、原纤蛋白及弹性纤维等。其中,弹性纤维是最主要的成分,约占皮肤质量的2%~4%,为皮肤、肺、大血管等富含弹力纤维的器官提供弹性。弹性纤维主要由弹力蛋白核心与微纤维构成。微纤维则由微纤毛构成,其被覆于弹力蛋白核心的外周,为弹力蛋白的沉积提供支架。微纤毛包括衰老关键蛋白fibrillin和微纤毛相关的糖蛋白等[6]。fibullins主要分布于微纤维的内表面,主要参与弹力蛋白沉积到微纤毛支架上及微纤维与细胞表面相互作用的过程[6]。
弹力蛋白由成纤维细胞及平滑肌细胞以弹力蛋白原的形式合成和分泌。弹力蛋白原交联过程在铜离子依赖的赖氨酰氧化酶(LOX)的介导下进行,其作为弹性纤维具备弹性及不溶性的基础。大部分弹力蛋白是在胎儿发育过程中累积的,少部分在出生后积累[7-8]。
以上过程中的任何环节出现异常,都将影响其下游的信号通路,从而影响细胞与弹性纤维的相互作用,最终导致皮肤弹性下降。
3 常染色体隐性遗传性皮肤松弛症的分型及特点
常染色体隐性遗传性皮肤松弛症(autosomal recessive cutis laxa,ARCL)是发病率最高且临床表现最为复杂的类型[9]。因其损害程度及受累器官变化多样,加之临床表现错综复杂,给临床的诊治造成巨大困难。笔者以下主要介绍ARCL最为常见的Ⅰ、Ⅱ、Ⅲ型的临床特点及其相关的分子生物学研究进展。
3.1 ARCL-Ⅰ型ARCL-Ⅰ型的主要临床表现包括典型重度肺气肿和致死性血管病变,其最常见临床表现包括肺不张、肺气肿、胃肠道憩室、泌尿生殖系统憩室及血管发育异常。血管发育异常主要表现为动脉瘤、纤维肌性动脉发育不良和狭窄等[10-11]。此外,颅骨异常、囟门闭合延迟、关节松弛及髋关节脱臼、腹股沟疝等也是常见临床表现[12-13]。学者们一致认可依据其致病基因,将其分为ⅠA(ARCL-ⅠA)、ⅠB(ARCL-ⅠB)及ⅠC(ARCL-ⅠC)三种[14]。研究发现,衰老关键蛋白4(FBLN4)和FBLN5基因突变常引起严重的肺气肿,但不伴血管迂曲或动脉瘤的形成[12];而人潜伏转化生长因子β结合蛋白4(LTBP4)基因突变患者的胃肠道及泌尿生殖道的临床症状更严重。
3.1.1 ARCL-ⅠAARCL-ⅠA是由Fibulin-5(FBLN5)突变导致的皮肤松弛症。Fibulin-5是Fibulin家族成员之一,Fibulin家族成员广泛分布于富含弹力蛋白的组织中,能直接与弹力蛋白结合,或者连接原纤蛋白的微纤维,从而在弹力纤维及细胞外基质的形成过程中发挥重要作用。其编码基因定位于人染色体的14q32.1,所编码蛋白是一种含有448个氨基酸的糖蛋白,分子质量约为60 ku。Fibulin-5作为细胞外基质蛋白,主要分布于大血管、心脏瓣膜、主动脉弓、肺、子宫及皮肤等含丰富弹性纤维的器官,因此,当Fibulin-5基因突变时,其他富含Fibulin-5的器官也会受累,从而表现出相应的临床症状。
3.1.1.1 临床表现ARCL-ⅠA主要临床特征为严重的发育性肺气肿,常常表现为致死性的新生儿呼吸窘迫,其他伴随症状包括主动脉瓣膜狭窄,肺动脉狭窄及腹股沟疝等。
3.1.1.2 分子生物学机制 研究发现Fibulin-5具有多种生物学功能,包括调控类赖氨酰氧化酶-1(LOXL1)、LOXL2、LOXL4、原纤蛋白-1、弹力蛋白原、LTBP2及LTBP4等分子的生物学功能,见图1[15]。以上分子与Fibullin-5结合不仅可促进弹力蛋白原凝聚及交联,同时有助于形成弹性纤维。此外,Fibulin-5突变后亦可影响蛋白的折叠及分泌功能,进而导致弹力蛋白不能与微纤毛整合。因此,当Fibullin-5基因突变致其功能受损时,Fibullin-5参与的上述过程均受到影响,最终导致皮肤松弛症的发生,见图2。
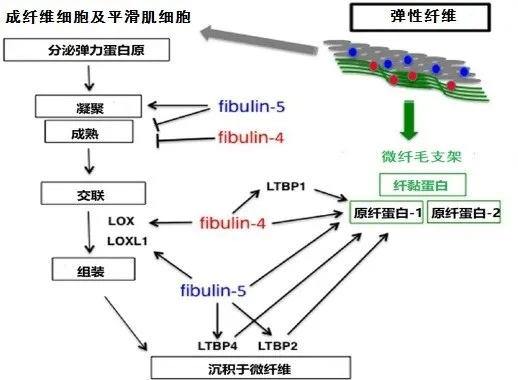
Fig.1 The pathogenesis genes involved in ARCL-typeⅠregulated the formation of elastic fibers图1 ARCL-Ⅰ型的各致病基因调控弹力纤维形成过程示意图
3.1.2 ARCL-ⅠBARCL-ⅠB的致病基因为Fibulin-4(FBLN4)。Fibulin-4与Fibulin-5属于同一家族成员,其编码基因定位于人染色体的11q13.1-q13.2区域,所编码蛋白是一种由442个氨基酸组成的细胞外基质蛋白,分子质量约50 ku。Fibulin-4不仅参与弹力纤维的形成过程,而且对于胶原的微纤毛形成过程具有重要的调控作用[12,16]。Fibulin-4两个等位基因部分或全部突变,均会因Fibulin-4蛋白水平的显著降低或缺如而引起严重的临床症状,大部分患者在出生后18个月内夭折,这一结论与体外实验研究结果一致[17]。Fibulin-4突变患者的临床症状依Fibulin-4蛋白功能受影响的程度而呈现多样性。
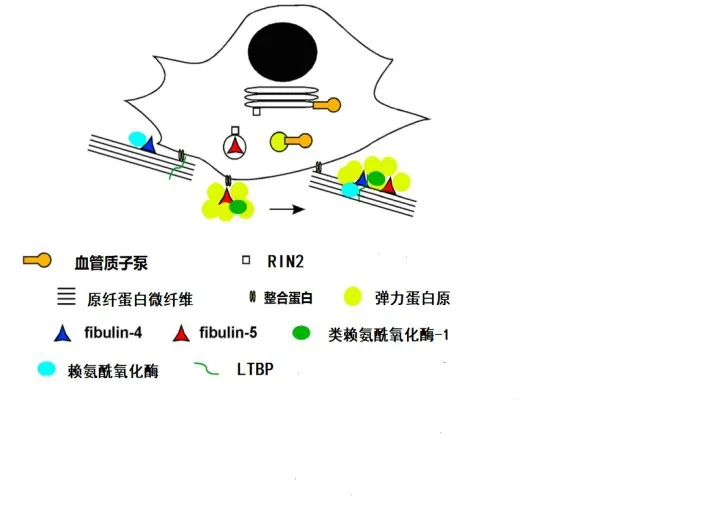
Fig.2 The molecular biological mechanism of regulating elastic fibers involved the ARCL-typeⅠ图2 ARCL-Ⅰ型的相关致病分子调控弹力纤维形成的分子生物学机制
3.1.2.1 临床表现FBLN4突变所致的ARCL-ⅠB可累及全身各系统,主要临床表现包括动脉迂曲、动脉瘤、肺动脉高压、发育性肺气肿、脆骨病、关节松弛、膈疝及腹股沟疝等[10,13]。
3.1.2.2 分子机制Fibulin-4可与弹力蛋白原、原纤蛋白-1及LTBP1相结合[18],通过影响这些分子,进而影响弹力纤维的形成。Fibulin-4与Fibulin-5的功能区分主要通过选择与不同的配体结合发挥各自调节作用。若两者均与同一种分子结合,则主要通过结合细胞类型的差异或亲和力的不同来确保两者在弹力纤维的组装过程中各司其职,互不替代(具体过程见图1)。例如,Fibulin-5对于弹力蛋白原的亲和力要高于原纤蛋白-1,而Fibulin-4则刚好相反,对原纤蛋白-1的亲和力要高于弹力蛋白原。Fibulin-4与弹力蛋白原、LOX形成三聚体,易化弹力蛋白的交联作用[19-20];而Fibulin-5则加强弹力蛋白原的凝聚作用[21]。Fibulin-4和Fibulin-5均调控弹性蛋白原凝聚成熟过程中的颗粒大小[21]。Fibulin-5缺乏可导致弹力蛋白的颗粒直径过大,且不能与微纤维支架整合[15,22](具体过程见图2)。与此相反,Fibulin-4突变则导致弹性纤维数量显著减少,表现为弹力蛋白的交联过程受阻及弹力纤维的结构紊乱[20],最终导致相应的临床症状(具体过程见图2)。
3.1.3 ARCL-ⅠCARCL-ⅠC也被称为Urban-Rifkin-Davis综合征,由LTBP4突变造成。LTBP4在3种不同启动子作用下产生3种亚型,1个小的LTBP4S及2个大的LTBP4L。在人类发现LTBP4突变时其三种亚型均受到影响,而在小鼠只发现LTBP4S异常。LTBP4S完全敲除小鼠的弹性纤维表型与人类完全缺乏的表现一致,比如发育性肺气肿及弹力纤维形态异常,即弹力蛋白表面光滑而无微纤毛[23]。但LTBP4突变造成的胃肠道及泌尿生殖道的症状目前只在人类观察到,在LTBP4S完全敲除的小鼠并未发现,这一现象提示消化系统及泌尿生殖道的发育可能主要依赖于LTBP4L[24]。
3.1.3.1 临床表现 严重的发育性肺气肿、憩室病、胃肠道的扩大和(或)狭窄、膀胱憩室、膈疝及腹股沟疝均是其常见的临床表现[25]。LTBP4突变的患者常死于呼吸衰竭及肠穿孔。大量研究证实LTBP4具备两种功能,一是调控转化生长因子(TGF)-β及其下游信号通路的作用,一是有助于弹性纤维的组装[26]。
3.1.3.2 分子机制LTBP4不同亚型的功能具有差异性,如LTBP4L偏向于与TGF-β结合,从而调控其下游信号通路;而LTBP4S则倾向于与细胞外基质的形成密切相关[27]。LTBP4可与Fibulin-5、原纤蛋白-1结合,从而有助于Fibulin-5与弹力蛋白形成的复合体锚定到微纤毛,见图1。LTBP4也被报道与纤黏蛋白及硫酸乙酰素蛋白多糖(HSPGs)相互结合,在细胞黏附过程中发挥着至关重要的作用[28]。人LTBP4编码C端细胞黏附区域的基因突变导致微纤毛束变厚和弯曲,提示LTBP4在调控细胞与细胞外基质分子沉积到弹力纤维的过程中具有至关重要的作用[25]。
3.2 ARCL-Ⅱ型
3.2.1 临床表现 该型的主要表现为广泛性皮肤松弛、皱褶,但面部症状通常较轻。特征性临床症状为前囟闭合延迟、前额突出、轻度尖颅、V型眉、眼裂下斜、龋齿、智力缺陷,常伴有宫内发育迟缓、臀部错位、鸡胸、脊柱侧凸等骨骼异常、腹股沟疝和扁平足[29-30]。依据其突变分为ⅡA(ATP6V0A2基因突变)和ⅡB(PYCR1基因突变)两型。
3.2.2 ARCL-ⅡAARCL-ⅡA由编码囊泡内质子泵V-ATP酶亚基的ATP6VOA2基因突变导致。ATP6VOA2基因突变导致N-连接和O-连接的糖基化异常,囊泡转运受阻,最终导致弹力蛋白原积聚于高尔基体,呈现多系统代谢性疾病。目前临床上可通过监测血浆中的转铁蛋白或载脂蛋白CⅢ的N-连接糖基化及O-连接糖基化水平,进而评估ATP6VOA2突变程度[31]。
3.2.2.1 临床表现ARCL-ⅡA的主要临床表现包括严重的神经系统缺陷,如巨脑回、小头畸形、肌无力、癫痫、近视、神经退行性病变及Dandy-Walker畸形。临床特征为全身广泛的皮肤松弛、囟门闭合延迟或不闭合、尖颅、关节过伸,同时具备神经系统异常症状及面部畸形的表现。典型的神经系统异常症状包括发育延迟、智力障碍、肌无力、小头畸形、失聪、癫痫,甚至头磁共振提示脑实质呈鹅卵石样发育不良。典型的面部畸形特征表现为前额突出、V型眉、眼裂下斜、长人中、面颊下垂及朝天鼻。此外,也可出现胎儿宫内发育迟缓、蓝色巩膜、漏斗胸、腹股沟疝、扁平足及先天性髋关节脱位等症状,部分患者亦可患有近视或斜视等眼部疾患。
3.2.2.2 分子机制 临床检测发现N-连接和O-连接的糖基化蛋白水平异常,即:含4个叶酸残基的转铁蛋白水平减少,而含2个或3个叶酸的转铁蛋白水平显著增加,以上生化指标的异常提示N-连接糖基化的蛋白水平显著增加。此外,包含2个叶酸残基的血浆载脂蛋白CⅢ减少,含单个叶酸残基的转铁蛋白含量增加,这两种现象均提示ATP6VOA2基因突变[32]。新生儿在围生期转铁蛋白水平可能处于正常范围内,但随着日龄增加,转铁蛋白异常的表现愈发明显。由此可见,生化检测是区分ARCL-ⅡA与ARCL-ⅡB的重要方法,对于ARCL-Ⅱ型的分型具有重要的指导意义。
3.2.3 ARCL-ⅡBARCL-ⅡB由线粒体酶PYCR1基因突变造成。该型主要由参与脯氨酸代谢的PYCR1基因突变导致线粒体功能异常[33]。PYCR1编码的酶主要功能是以NAD(P)H-依赖的方式将5-羧酸二氢吡咯转变为脯氨酸[34],从而影响线粒体的能量代谢过程。该型常见的临床表现为腹部及背部的皮肤松弛、先天性髋关节脱位、胎儿宫内或生后生长发育迟缓、骨质疏松、三角形面容或早衰面容、蒜头鼻、下颌突出、内眦赘皮、蓝色巩膜,大耳及小头畸形等。神经系统表现为肌无力、发育迟缓、智力障碍、胼胝体发育不全或缺如,极少数可能智力正常。
3.3 ARCL-Ⅲ型ARCL-Ⅲ型,又称为De Barsy综合征,该型主要由乙醛脱氢酶18家族成员A1(ALDH18A1)基因突变造成[35-36]。ALDH18A1编码的蛋白主要功能是将谷氨酸转变为脯氨酸、鸟氨酸及精氨酸的生物合成过程中的中间代谢产物5-半醛谷氨酸。若ALDH18A1基因突变,则脯氨酸、鸟氨酸及精氨酸三种氨基酸的生物合成过程都将受到影响,因此,多系统都将受累并表现出相应的临床症状。该型主要以头发稀少、角膜异常、胎儿宫内发育迟缓和皮肤松弛等早衰外观为特征。患者出生时表现为皮肤薄、半透明、多皱褶和无弹性并伴有明显的可见静脉。眼睛特征性变化为角膜鲍氏膜弹性纤维降解导致角膜混浊[36]。大部分可同时伴有神经及骨骼系统的异常,包括认知和(或)语言功能受损、髋关节脱位、关节过伸、脊柱侧弯和足部畸形。部分患者与Ⅱ型有重叠的特征,但肌张力障碍和角膜异常提示为本型。
笔者总结了遗传性皮肤松弛症的各分型及其临床特征、致病基因及功能,见表1。皮肤松弛症的预后及干预措施因其遗传类型的不同而差异显著。常染色体显性遗传性皮肤松弛症(autosomal dominant cutis laxa,ADCL)和X连锁遗传性皮肤松弛症(X-linked cutis laxa,XLCL)根据患者的临床表现及其家族史较易鉴别诊断。ARCL因常伴严重的心血管系统损害,患儿多于儿童期死亡。与ADCL和XLCL相比,ARCL更依赖于有效的产前指导及咨询以降低其发病率。随着医学研究的发展及转化医学的广泛应用,可通过检测某些疾病的致病基因或相关蛋白以指导疾病的早期诊断及治疗,如目前通过生化检测转铁蛋白水平区分ARCL-ⅡA与ARCL-ⅡB等。因此,探究与疾病相关的基因及信号通路来指导ARCL的防治及早期诊断值得进一步研究探讨。
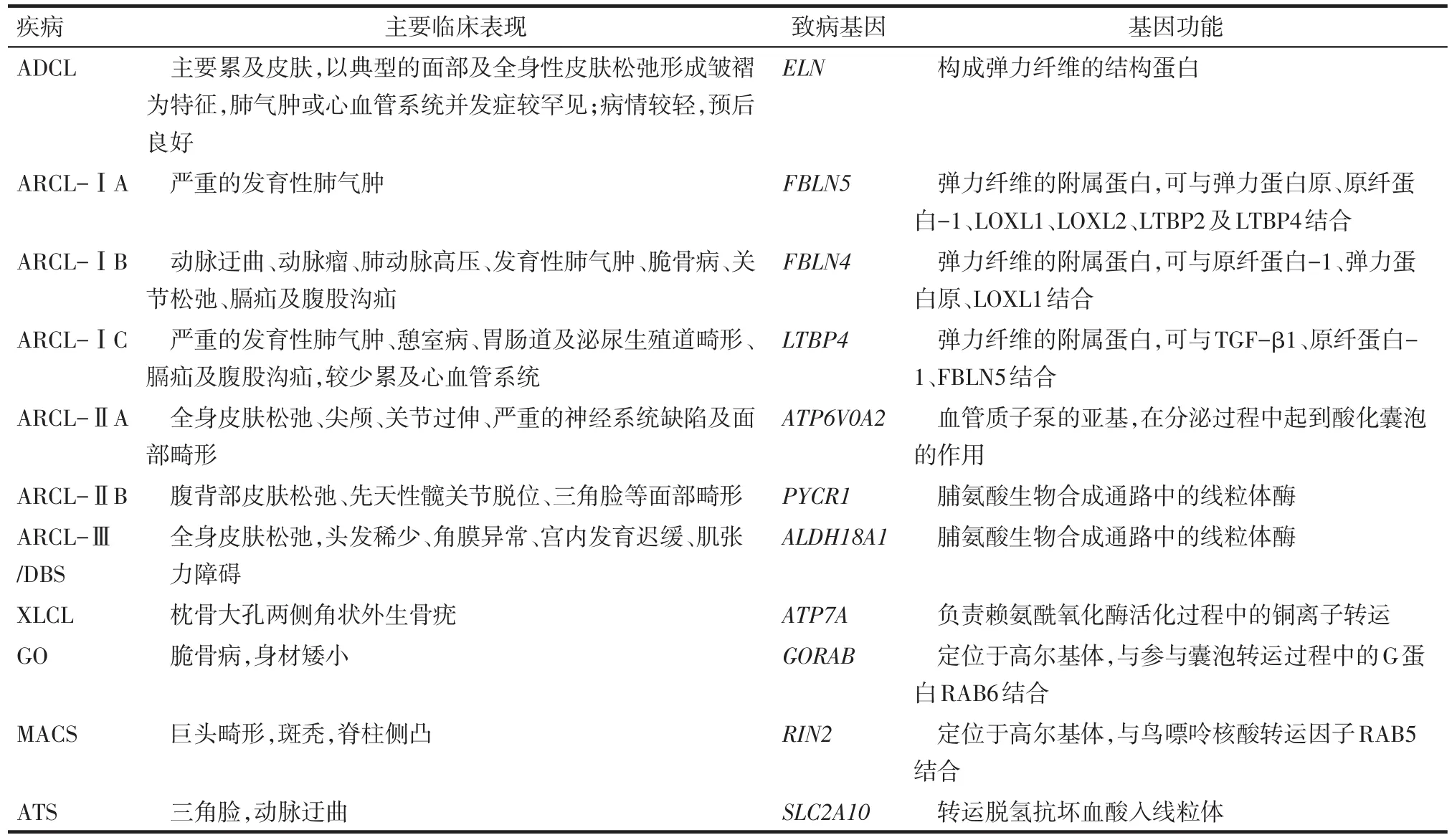
Tab.1 The specific manifestations of common types of cutis laxa and the corresponding genes and functions表1 常见皮肤松弛症的类型及其特征性的临床表现、相应的致病基因及其功能
猜你喜欢
英语世界(2023年6期)2023-06-30 06:29:10
中国现代医生(2022年19期)2022-11-04 10:13:29
昆明医科大学学报(2022年4期)2022-05-23 13:04:50
基层中医药(2021年8期)2021-11-02 06:24:54
中老年保健(2021年3期)2021-08-22 06:50:32
中国生殖健康(2020年2期)2021-01-18 02:51:26
小学生导刊(2018年13期)2018-06-29 03:49:00
西南军医(2015年1期)2015-01-22 09:08:34
河南医学研究(2014年5期)2014-02-27 14:52:41
儿童故事画报(2013年3期)2013-06-24 05:40:30
