
应用MiSeq测序分析自然发酵豆酱酱块中 微生物的多样性
2018-09-13 11:09解梦汐安飞宇武俊瑞唐筱扬乌日娜
食品工业科技 2018年16期
姜 静,解梦汐,安飞宇,武俊瑞,唐筱扬,乌日娜
(沈阳农业大学食品学院,辽宁沈阳 110866)
豆酱是一种传统的发酵豆制品,在中国作为调味剂已有几个世纪之久,在日本被称为Miso(味噌)[1],在韩国则叫做Doenjang[2]。作为一种传统的发酵食品,豆酱具有自己独特的色、香、味、形,长期以来深受我国各地人们的喜爱[3]。除此之外,豆酱中还含有许多对人体有益的生理活性物质,具有一定的保健功能[4],如抑制血清胆固醇、抑制脂肪肝的形成,预防肝细胞癌[5-6],抑制血管紧张素转化酶[7],降血压、清除放射性物质和抗癌[8-10]等。
传统发酵豆酱的生产过程主要分为两个阶段,前制曲阶段和后发酵阶段[11],其中制曲是影响豆酱产品质量的重要工艺[12],本身具有非常复杂的微生物生态系统,而这些微生物负责水解酱块发酵过程中的主要成分,包括蛋白质、脂肪和碳水化合物等,从而产生各种代谢产物,例如氨基酸、有机酸、活性代谢产物和苷元,对豆酱的营养、口感、风味和功能具有重要作用[13]。目前,工业制曲大多数厂家添加米曲霉(Aspergillusoryzae)和黑曲霉(Aspergillusniger)[14]。然而,添加单一菌种制曲使得豆酱的风味和口感远不如传统发酵豆酱。因此,研究传统发酵豆酱酱块中微生物多样性是十分重要的。
当前,豆酱制品酿制过程中的主要微生物可分为三大群体,即霉菌、细菌和酵母菌[15]。Lee等[16]采用DGGE技术分析豆酱中的微生物,发现豆酱中主要细菌是粪肠球菌(Enterococcusfaecalis),主要真菌是伞枝犁头霉菌(Absidiacorymbifera)、曲霉菌(Aspergillus)、假丝酵母菌(Candida);田甜[17]利用焦磷酸测序技术对东北豆酱自然发酵过程中微生物进行分析,结果表明,优势细菌为葡萄球菌属(Staphylococcus)和明串球菌属(Leuconostoc),优势真菌为赤霉菌属(Gibberella)、拟青霉属(Paecilomyces)和青霉菌属(Penicillium)。
近年来,随着基因测序技术的不断进步,越来越多的研究者利用Miseq第二代测序技术分析复杂环境微生物。相对于传统的纯培养法和第一代测序技术或酶切法,Miseq测序技术不仅可以产生测序覆盖深度更大的数据量,而且可以发现低丰度的细菌菌群。通过MiSeq测序技术分析传统发酵豆酱酱块中的微生物多样性,深入了解传统发酵豆酱酱块微生物群落结构,挖掘丰富的微生物资源并确定酱块中优势菌群,为工业化人工接种法生产酱类提供理论指导。
1 材料与方法
1.1 材料与仪器
6份成熟酱块样品 采集于沈阳胡台不同农家,编号分别为HT1、HT2、HT3、HT4、HT5、HT6,采集的样品于-20 ℃保存。
Eppendorf5417高速冷冻离心机 德国艾本德公司;Agilent 2100生物分析仪 美国Agilent公司;MiSeq测序仪 美国Illumina公司;Qubit 2.0核酸蛋白定量仪 美国Carlsbad公司;MetaVxTM试剂盒 美国Genewiz公司。
1.2 实验方法
1.2.1 DNA的提取 pH7.0、浓度为0.1 mol/L磷酸盐缓冲液悬浮8 g酱块样品,加入玻璃珠,振荡器振荡3 min;收集上清液弃去沉淀,洗涤三次;10000 r/min高速离心5 min,取上清液,得到菌体;收集的菌体用pH7.0、浓度为0.1 mol/L磷酸盐缓冲液悬浮,9000 r/min 5 min,洗涤三次;洗净的菌体悬浮在磷酸盐缓冲液中,用枪吹打后涡旋振荡均匀。采用Fast Prep结合CTAB法进行总DNA的快速提取[18]。
1.2.2 MetaVxTM文库构建和Illumina MiSeq测序 以30~50 ngDNA为模板,PCR引物扩增原核生物16S rDNA上V3/V4区,扩增体系为20 uL,4 uL 5*FastPfu缓冲液,2 uL 2.5 mmol/L dNTPs,0.8 uL引物(5 umol/L),0.4 uLFastPfu聚合酶;10 ng DNA模板。反应参数:预变性条件为95 ℃,3 min;循环为95 ℃,30 s、55 ℃,30 s、72 ℃,30 s,27个循环;最后在72 ℃下延伸,10 min,4 ℃保存。采用包含“CCTACGGRRBGCASCAGKVRVGAAT”序列的上游引物和包含“GGACTACNVGGGTWTCTAATCC”序列的下游引物扩增V3和V4区,采用包含“GTGYCAGCMGCCGCGGTAA”序列的上游引物和包含“CTTGTGCGGKCCCCCGYCAATTC”序列的下游引物扩增V4和V5区。另外,通过PCR向16S rDNA的PCR产物末端加上带有Index的接头,以便进行NGS测序。
使用生物分析仪检测文库质量,并且通过核酸蛋白定量仪检测文库浓度。DNA文库混合后,按测序仪器使用说明书进行2×300 bp双端测序(PE),由MiSeq自带的MiSeq Control Software(MCS)读取序列信息。
1.2.3 ITS区文库构建和 Illumina MiSeq 测序 以50~100 ng DNA为模板,PCR扩增真菌ITS区。采用包含“ACCTGCGGARGGAT”序列的上游引物和包含“GAGATCCRTTGYTRAA”序列的下游引物扩增ITS1区,采用包含“GTGAATCATCGARTC”序列的上游引物和包含“TCCTCCGCTTATTGAT”序列的下游引物扩增ITS2区。再通过PCR向ITS区的PCR产物末端加上带有Index的接头,以便进行NGS测序。
1.3 数据分析
Miseq测序结果为双端测序得到的正反向reads首先进行两两组装连接,过滤拼接结果中含有N 的序列,保留序列长度大于200 bp的序列。经过质量过滤,去除嵌合体序列,最终得到的序列用于OTU分析,使用VSEARCH(1.9.6)进行序列聚类(序列相似性设为 97%),比对的16S rRNA和18S rRNA参考数据库是Silva 119。然后用RDP classifier贝叶斯算法对OTU的代表性序列进行物种分类学分析,并在不同物种分类水平下统计每个样本的群落组成。基于OTU的分析结果,采用对样本序列进行随机抽样的方法,分别计算 Shannon、Chao等α多样性指数,并作出稀释曲线。通过 PCA分析比较样本的微生物群落间是否有显著差异。
2 结果与分析
2.1 样品测序数据
如表1所示,不同农家酱块中真菌和细菌菌群α-多样性分析。可操作分类单元(OTU)是指根据某一人为设定的序列相似度阈值,将来自一个或多个样本的序列进行归并,彼此间相似度高于该阈值的序列都将归并为一个OTU;物种丰富度指数(ACE)是用来估计群落中含有OTU数目的指数;辛普森多样性指数(Simpson)是用来定量的描述一个区域的生物多样性;赵氏指数(Chao)是反映样品中群落的丰富度;香农指数(Shannon)用来估计样品中微生物多样性;覆盖率(Coverage)是指各样本文库的覆盖率,其数值越高,则样本中序列没有被检测出的概率越低。

表1 每个样本的多样性指数Table 1 Diversity estimates for each sample
测序6个样本ACE多样性指数变化范围在22~34.5之间,Simpson指数变化范围在0.03~0.79,各样品的Shannon指数变化范围在0.14~2.87,Chao指数变化范围在22~33,表中97%的物种型的样本文库覆盖率为1,这表明样品中菌群型基本被鉴定出来。
2.2 稀释曲线
采用对测序序列进行随机抽样的方法,以抽到的序列数与它们所能代表OTU的数目构建曲线,即稀释性曲线,如图1、图2所示。

图1 细菌的稀释曲线Fig.1 Rarefaction curve analysis of bacteria

图2 真菌的稀释曲线Fig.2 Rarefaction curve analysis of fungi
每个样品的取样深度,从6份样品中随机抽取的测序条数如图1横轴所示,纵轴则表示基于该测序条数能构建的OUT数量,在<6000条序列时,OTU值随着序列增加而迅速增加;在>6000条序列时,OTU数增加缓慢,曲线趋于平坦时,说明测序数据量合理,更多的数据量对发现新OTU的边际贡献很小。该分析是基于OTU序列差异水平在0.03即相似度97%的水平上进行运算的。
2.3 样品中细菌群落组成分析
采用RDP classifier贝叶斯算法对97%相似水平的OTU代表序列进行分类学分析,并在门和属水平上统计每个样品的群落组成。
从图3细菌在门水平上的分布来看,6份样品中共检测出4个已知的细菌门,分别为厚壁菌门(Firmicutes)、变形菌门(Proteobacteria)、放线菌门(Actinobacteria)和蓝藻菌门(Cyanobacteria),其中优势菌门均为厚壁菌门(Firmicutes),所占比例在81.9%~99.9%之间。

图3 细菌在门水平群落结构Fig.3 Bacterial community structure at phylum level
如图4所示,从细菌在属水平的分布来看,6份样品共检测出23个已知属。其中主要细菌为乳杆菌属(Lactobacillus)、肠球菌属(Enterococcus)、肉杆菌属(Carnobacterium)和明串珠菌属(Leuconostoc),但在各个样品中,优势菌属存在显著差异。例如,样品HT3(73%)、HT5(47.8%)中优势菌属为乳杆菌属(Lactobacillus);样品HT1(55%)、HT6(53.1%)优势菌属为肠球菌属(Enterococcus);样品HT4(40.8%)优势菌群是明串株菌属(Leuconostoc);样品HT2(41.2%)优势菌属为肉杆菌属(Carnobacterium)。
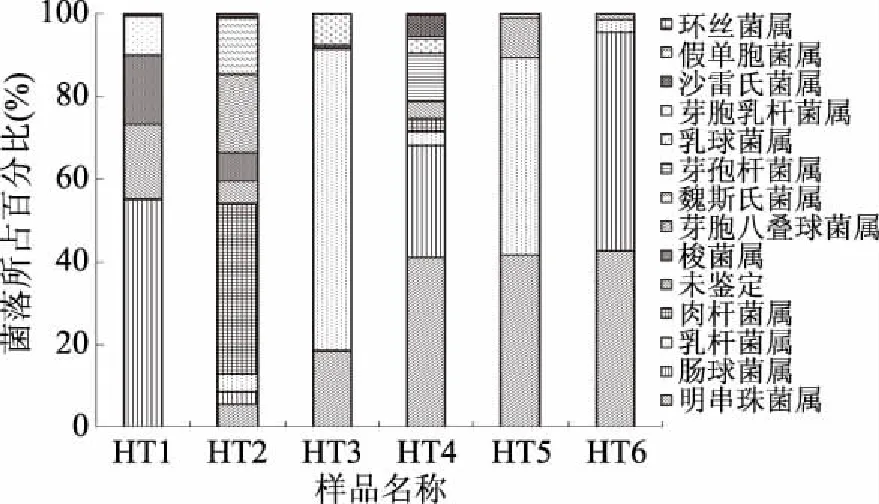
图4 细菌在属水平群落结构Fig.4 Bacterial community structure at genus level
明串珠菌(Leuconostoc)是革兰氏阳性、耐氧的一种乳酸细菌,是美国FDA公认的安全菌株(GRAS),是韩国泡菜发酵过程中起主要作用的微生物[19]。Nam等[20]在韩国大酱中检测出肠膜明串珠菌(L.mesenteroides)和L. sp. HBB8。在正常情况下,肠膜明串珠菌(L.mesenteroides)产乳酸,可以在高盐和高糖环境中生长[21]。在本研究中,明串珠菌(Leuconostoc)存在于除HT1以外所有样品中,可以推测明串珠菌(Leuconostoc)应该是酱块中的主要细菌之一,对豆酱的发酵可能起重要作用。
乳杆菌(Lactobacillus)存在于所有样品中(HT1<0.02%),是HT3和HT5的优势菌种。武俊瑞等[22]初步推断嗜盐四联球菌(Tetragenococcushalophilus)和植物乳杆菌(Lactobacillusplantarum)是黑龙江传统发酵豆酱中优势乳酸菌菌群。植物乳杆菌(Lactobacillusplantarum)广泛存在于酱油和豆酱中,它是FDA批准使用的乳酸菌[23]。由于酱块是自然发酵豆酱发酵过程中的重要来源之一,所以有理由认为酱块中的乳杆菌主要是植物乳杆菌(Lactobacillusplantarum)。植物乳杆菌(Lactobacillusplantarum)可以降解杂醇为组氨酸、精氨酸和天冬氨酸,对豆酱风味有着重要作用。植物乳杆菌(Lactobacillusplantarum)不仅有利于食品发酵,改善风味,提高营养价值,而且作为益生菌存在于人体内对人体具有有益的调节作用,如改善肠道功能、抗肿瘤、增强免疫力、抗氧化、降低胆固醇、降低亚硝酸盐[24-25]。
酱块中肠球菌(Enterococcus)是相对丰富的细菌菌群,在本研究中,肠球菌(Enterococcus)存在于所有样品中,且半数样品中肠球菌(Enterococcus)所占比例超过27.1%。Jeong等[26]发现在酱块发酵过程中,随着芽孢杆菌(Bacillus)数量增多肠球菌(Enterococcus)数量降低。事实上,样品HT1和HT6中肠球菌(Enterococcus)分别占55%和53.1%,而芽孢杆菌分别占0.02%和0.04%,这个可以解释为肠球菌(Enterococcus)在豆酱发酵过程中起重要作用,它可以阻止某些细菌例如芽孢杆菌(Bacillus)的生长[27]。
2.4 样品中真菌群落组成分析
如图5真菌在门水平上的分布来看,6份样品已知真菌主要包括子囊菌门(Ascomycota)、接合菌门(Zygomycota)和担子菌门(Basidiomycota)。在当前的研究中,6份样品中子囊菌门(Ascomycota)均占主导地位,所占比例在50.8%~99.9%之间。
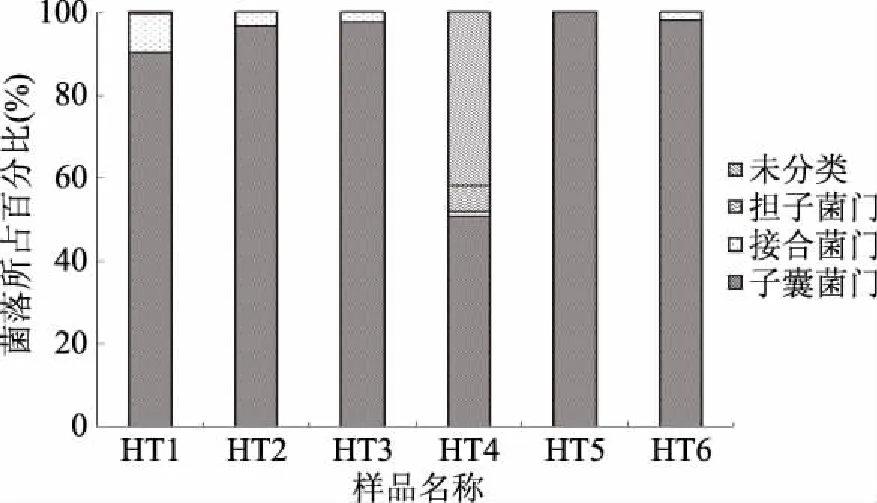
图5 真菌在门水平群落结构Fig.5 Fungi community structure at phylum level
真菌在属水平分类,如图6所示,6份样品归属于13个已知属。主要检测出来的真菌有青霉菌属(Penicillium)、念珠菌属(Candida)、毛霉菌属(Mucor)等,但各个样品间优势菌群存在差异。样品HT1、HT3、HT6中优势真菌为青霉菌(Penicillium、78.7%~97%);样品HT2优势菌群是念珠菌(Candida,61.1%);样品HT4、HT5占比较高的菌群均是尚未分类菌群。

图6 真菌在属水平群落结构Fig.6 Fungi community structure at genus level
真菌在豆酱发酵过程起重要作用,它可以将大分子营养物质降解为小分子[28]。目前对酱块中细菌的研究非常多,但对真菌菌群的研究相对较少。Hong等[29]对韩国酱块中菌群进行研究,发现曲霉菌(Aspergillus)、毛霉菌(Mucor)、青霉菌(Penicillium)、帚霉菌(Scopulariopsis)等为韩国酱块中的主要真菌。Jung等[30]对传统韩国酱块中微生物群落动态进行了分析,发现在整个发酵过程中,主要检测出来的真菌为地丝菌属(Geotrichum)、帚菌属(Scopulariopsis)、红曲霉属(Monascus)、镰刀菌属(Fusarium)和曲霉属(Aspergillus)。而在本研究中,青霉菌(Penicillium)和毛霉菌(Mucor)存在于所有样品中,是样品中的优势菌群。由于温度和水分等是影响真菌群落的重要因素,所以酱块中真菌群落组成会有差异。
2.5 主成分分析
PCA作为一种无监督的分析方法,反映数据的原始状态,观察试验样品的自然分布和组别关系[31]。本研究对6份酱块真菌和细菌进行了PCA分析,如图7所示(细菌主成分得分图),利用各样品序列间的进化信息计算样品距离,观察6份不同酱块样品多样性差异,其中第一主成分(PC1)和第二主成分(PC2)可以解释59.93%的原变量信息。PCA分析中有些站点聚在一起,有些距离相对较远,故可将6份样品细菌群落大致分为3组:组1为样品HT1、HT3、HT5、HT6;组2 为样品HT2;组3为样品HT4,表明大多数样品细菌菌群之间存在密切的联系。因此可以看出,同一地区制作的酱块,绝大多数细菌群落仍具有紧密联系。

图7 细菌主成分得分图Fig.7 Bacterial PCA scoring diagram
如图8所示(真菌主成分得分图),第一主成分(PC1)和第二主成分(PC2)可以解释66.39%的原变量信息。PCA得分图显示将6份酱块样品真菌群落大致分为3组:组1为样品HT1、HT2、HT3、HT5;组2为样品 HT4;组3为HT6。其中HT1、HT2、HT3、HT5显示真菌之间有密切的联系,说明真菌群落在同一地区制作的酱块中变化不显著。
3 结论与讨论
在本研究中,利用Miseq测序技术对采自中国辽宁胡台不同农家的6份酱块微生物群落结构进行分析。结果表明,在门水平上,优势细菌为厚壁菌门(Firmicutes),真菌为子囊菌门(Ascomycota);在属水平上,主要细菌为乳杆菌属(Lactobacillus)、肠球菌属(Enterococcus)、明串珠菌属(Leuconostoc),主要真菌为青霉菌属(Penicillium)和毛霉属(Mucor)。
张颖等[32]通过PCR-DGGE技术结合微生物多样性测序方法分析豆酱发酵过程中细菌的多样性,结果表明,肠球菌属(Enterococcus)、四联球菌属(Tetragenococcus)和乳杆菌属(Lactobacillus)为豆酱样品不同发酵阶段的优势细菌菌属。韩国学者Kim[33]利用454焦磷酸测序对韩国豆酱细菌菌群进行分析,结果表明韩国豆酱主要细菌是乳酸菌,包括嗜盐四联球菌(Tetragenococcushalophilus)、肠球菌(Enterococcus)、明串珠菌(Leuconostoc)、乳杆菌(Lactobacillus)。在本研究中,乳酸菌中除嗜盐四联球菌(Tetragenococcushalophilus)外,其他优势细菌基本符合上述研究。嗜盐四联球菌(Tetragenococcushalophilus)是豆酱中的主要乳酸菌,但有趣的是,先前运用分子生物学方法均没有在酱块中发现嗜盐四联球菌(Tetragenococcushalophilus)[34],豆酱的制作就是将酱块和盐水按一定比例混合,发酵成为豆酱。因此可以猜测,嗜盐四联球菌(Tetragenococcushalophilus)存在于混合过程中的盐水中或者在酱块中处于休眠状态且含量极低。
刘春凤等[18]利用基因克隆文库构建技术对发酵成熟酱醅微生物进行分析,主要真菌为接合酵母属(Zygosaccharomyces)、Rhizochaete属、曲霉属(Aspergillus)、红酵母属(Rhodotorula)。韩国学者Hong等[35]利用免培养法检测韩国酱块中的真菌,发现曲霉菌(Aspergillus)、帚霉菌(Scopulariopsis)、毛霉菌(Mucor)和青霉菌(Penicillium)为韩国酱块中的主要真菌。本研究中的优势菌群为青霉菌(Penicillium)和毛霉菌(Mucor),由于酱块中的水分和发酵时的温度是决定真菌菌群的重要因素,所以不同地区、不同手法制作的酱块,优势菌群之间存在差别。
本文中除了真菌和细菌的优势菌群外,其余大多数是占比重小于1%的群落参与酱块的发酵,它们占总菌群种类的80%以上。虽然不能解释这些占比重小于1%的微生物是如何与优势菌群相互作用,但这些微生物可能在维持微生物群落稳定上起重要作用,在适当的条件下,它们有潜力成为主要的微生物来源[20]。因此,酱块不仅由几个主要微生物种群组成,还包括许多占比重小的微生物群落。
猜你喜欢
中国调味品(2022年9期)2022-08-30
昆明医科大学学报(2022年2期)2022-03-29
餐饮世界(2021年10期)2021-11-20
食品安全导刊(2021年20期)2021-08-30
河南科学(2020年3期)2020-06-02
国际呼吸杂志(2019年1期)2019-01-28
中国调味品(2017年2期)2017-03-20
现代检验医学杂志(2016年2期)2016-11-14
环境科技(2016年3期)2016-11-08
中国酿造(2016年12期)2016-03-01
