
Potential therapeutic interventions based on the role of the endoplasmic reticulum stress response in progressive neurodegenerative diseases
2018-09-11 07:26:50BasantK.Puri,GerwynMorris
中国神经再生研究(英文版) 2018年11期
In 1945, Porter et al. published an electon microscopy study of cultured chick fibroblasts in which they observed:‘a granular background and details of a darker lacelike reticulum which in places appears to be made up of chains of “vesicles”’ (Porter et al., 1945). This constituted the first published observation of the endoplasmic reticulum (ER) and, while it was not evident at that time, this cytoplasmic system of interconnecting membrane-lined channels, comprising vesicles, tubules and cisternae, has numerous important functions.
Role of the ER: The ER can be divided into rough ER(RER), in which ribosomes are attached to the cystolic surface, transitional ER (tER), in which peptides and proteins are packaged for transportation to the Golgi apparatus, and smooth ER (SER), which is devoid of ribosomes. Major ER functions include: biosynthesis, folding and transportation of proteins/peptides; biosynthesis and transportation of phospholipids and steroid molecules(including steroid hormones); storage and release of calcium ions; detoxification; ER-associated degradation(ERAD) of misfolded or unfolded proteins/peptides; glycosylation; and carbohydrate metabolism (Agostinis and Samali, 2012).
Unfolded protein response (UPR): The mechanisms described in this section are illustrated in Figure 1. Inadequate ERAD activity is associated with a build-up of misfolded or unfolded proteins/peptides in the ER.In turn, this is associated with an adaptive or protective ER homeostatic stress response named the UPR. ERAD activity may be insuf ficient as a result of excess reactive oxygen species synthesis, reduced anti-oxidant efficiency or disturbed calcium ion homeostasis. The accumulated misfolded or unfolded proteins/peptides become bound to the master ER chaperone glucose-regulated protein 78(GRP78), which in turn may activate the following three ER transmembrane protein stress sensors which are involved in mediating the UPR: protein kinase RNA-like endoplasmic reticulum kinase (PERK); inositol-requiring enzyme 1α (IRE1α); and activating transcription factor 6(ATF6).
Activation of PERK initially causes dimerisation and autophosphorylation of PERK receptors. This is then followed by adaptive signalling cascades which lead to changes in gene expression relating to redox control, including in respect of antioxidant enzymes involved in the detoxification of reactive oxygen species (ROS).
IRE1α activation is also followed by dimerisation and autophosphorylation. The subsequent adaptive signalling cascades lead to changes in the expression of genes related to ERAD, ER chaperones and lipid biosynthesis, as well as to activation of p38 mitogen-activated protein kinase(MAPK) and c-Jun N-terminal kinase (JNK) which in turn are associated with mechanisms relating to cell death and cell survival.
Activated ATF6 is transported to the Golgi complex.Here, following protease cleavage reactions, active ATF6 fragments then enter the nucleus, via nuclear pores, and cause changes in the expression of genes related to ERAD,ER chaperones and lipid biosynthesis.
Morris et al. (2018a) have recently reviewed the pathophysiological role of the ER in progressive neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, multiple sclerosis, amyotrophic lateral sclerosis,bipolar disorder, major depressive disorder and schizophrenia. They point out that the UPR is associated with the development of chronic inflammation, through activation of all three types of the above ER transmembrane protein stress sensors. For example, PERK activation can set in motion a feedforward loop of increasing inflammation (indeed, such activation in astrocytes can initiate neuroinflammation), while ATF6 activation leads to essentially pro-inflammatory actions and upregulation of macrophage toll-like receptor activity. UPR activation is also associated with the development of oxidative and nitrosative stress, which can take place via the following mechanisms: increased ROS production upregulation of protein disulphide isomerase, upregulation of ER oxidative protein folding, glutathione oxidation, increased protein S-nitrosylation, and increased calcium ion ef flux into the mitochondria from the ER (Morris et al., 2018a).
ER-mitochondria interrelationship: The ER and mitochondria interact in a precise, controlled fashion(Agostinis and Samali, 2012; Morris et al., 2018a). The interactions are reflected anatomically by the existence of mitochondria-associated membranes (MAMs), which are specific ER membrane subdomains which interact with the outer mitochondrial membrane (OMM). They were first specifically described in a crude rat liver mitochondrial fraction by Jean Vance in 1990 (although there was evidence of ER-OMM interactions from two to three decades earlier) and appeared to be involved in the biosynthesis and transfer, between the ER and mitochondria,of phospholipids (Vance, 1990). Since then, it has become clear that they have other roles including, notably, involvement in calcium ion cross-talk between the ER and mitochondria. Furthermore, the way in which the ER is tethered to the OMM at the MAM domain is complex and involves numerous different proteins.
Calcium ion cross-talk between these two organelles has a crucial role to play in the phenomenon of mitochondrial outer membrane permeabilisation (MOMP).
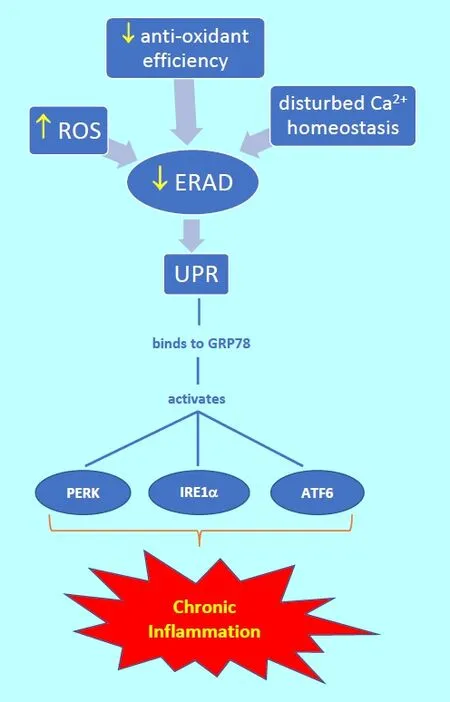
Figure 1 Diagrammatic summary of endoplasmic reticulum (ER)processes leading to chronic in flammation.
In particular, ER stress may be associated with changes in this calcium ion cross-talk, which in turn may cause MOMP. As a consequence, cytochrome c, which is normally found associated with the inner mitochondrial membrane (IMM), where, for example, it is part of the electron transport chain (ETC), is released and thereby initiates apoptosis. Thus ER stress can be associated with mitochondrial changes and autophagy. It is therefore not surprising that ER stress may be associated with neurodegenerative disorders.
ER stress and progressive neurodegenerative diseases:Morris et al. (2018a) have marshalled evidence showing that the response of prolonged ER stress to chronic pathophysiological insults, with its attendant UPR overactivation, neuroinflammation, oxidative and nitrosative stress, and dysregulated calcium ion homeostasis,working through mitochondrial pathways, may lead to diminished neuronal resilience, dysfunction of synaptic activity and apoptosis, thereby contributing to progressive neurodegenerative diseases. Indeed, from a pathophysiological viewpoint, misfolded protein accumulation is of key importance in disorders such as Huntington’s disease (chorea) and amyloidosis. Furthermore, sub-lethal ER stress, leading to chronic UPR upregulation, appears to be of importance in the neuropathology of several progressive neurodegenerative diseases, including Alzheimer’s disease, multiple sclerosis, Parkinson’s disease and amyotrophic lateral sclerosis (also known as motor neurone disease or Lou Gehrig’s disease), and perhaps even psychiatric disorders such as bipolar mood disorder,major depressive disorder and schizophrenia (Morris et al., 2018a).
Importantly, there is evidence to suggest that inhibition of UPR pathways may be associated with neuroprotection. This naturally leads us to a discussion of potential therapeutic interventions for progressive neurodegenerative disorders.
Potential therapeutic interventions: Interventions which target ER stress and UPR pathways would offer new therapeutic targets for progressive neurodegenerative disorders, and have been described by Morris et al. (2018a).They include melatonin, coenzyme Q10(CoQ10) and N-acetylcysteine (NAC). We shall consider each of these in turn.
The pineal hormone melatonin (or N-acetyl-5-methoxy tryptamine), which is biosynthesised also in some non-pineal tissues and occurs naturally in certain foods, readily crosses the blood-brain barrier and its selective entry into mitochondria is aided by OMM glucose transporter 1 (Glut-1) or peptide 1 and 2 transporter proteins. It has antioxidant actions; these are possibly direct actions but indirect antioxidant actions may be important, for example through an increase in the activity of glutathione peroxidase and γ-glutamylcysteine synthetase. Actions of melatonin which might be therapeutic in the context of ER stress include: mitophagy regulation; restoration of calcium ion homeostasis; reduced mitochondrial oxidative stress; improved efficiency of the generation of adenosine triphosphate (ATP) by the ETC; reduced mitochondrial nitric oxide synthase expression; and reduced release of cytochrome c from the IMM (Morris et al.,2018a). At doses two to three orders of magnitude greater than the physiological doses that affect the circadian rhythm, melatonin appears to be able to prevent nitrosative and oxidative stress-induced mitochondrial dysfunction in non-human mammalian models of Alzheimer’s disease, Parkinson’s disease and Huntington’s disease; it has therefore been suggested that human clinical trials in these neurodegenerative diseases should be carried out,perhaps using daily doses of up to 100 mg oral melatonin(Cardinali et al., 2013).
CoQ (or ubiquinone) plays a key electon carrier role in the mitochondrial ETC, accepting electrons from complex I (NADH-Q oxidoreductase) and Complex II (succinate dehydrogenase) and forwarding them to Complex III (the cytochrome c reductase complex). It appears to have free radical scavenging properties and restores endogenous antioxidant activity, and it may protect against lipid peroxidation. It may also help downregulate the expression of genes encoding UPR-associated proteins, stabilise the mitochondrial permeability transition pore, and reduce levels of pro-in flammatory cytokines and ROS (Morris et al., 2018a). CoQ10supplementation at a daily dose of 500 mg for 12 weeks has been found to ameliorate markers of in flammation and oxidative stress in a randomised, double-blind, placebo-controlled trial of patients with relapsing-remitting multiple sclerosis (Sanoobar et al., 2015).There may be particular benefit from covalently bonding CoQ10to a lipophilic cation in order to achieve much higher mitochondrial matrix CoQ10concentrations than those possible from supplementing with ordinary CoQ10;indeed, preliminary evidence from studies of mitochondrial-targeted CoQ10(MitoQ), in which the cation moiety is triphenylphosphonium, shows promise in Parkinson’s disease (Snow et al., 2010). It has also been suggested that CoQ10may be therapeutically useful in mood disorders.
NAC is a precursor of L-cysteine and therefore of glutathione. NAC supplementation in mammalian studies,including in humans, has confirmed that NAC has anti-inflammatory, antioxidant and cytoprotective actions,with increased glutathione, reduced ROS, reduced lipid peroxidation, restored calcium ion homeostasis, reduced entry of calcium into mitochondria, increased mitochondrial membrane potential, increased OMM stability and increased efficiency of ATP production being reported(Morris et al., 2018b). It is particularly noteworthy that NAC supplementation has been shown to mitigate ER stress levels in a dose-dependent manner in mammalian experiments (Liu et al., 2008; Machado et al., 2014).Currently, NAC is being actively studied as a potential therapeutic agent in a range of progressive neurodegenerative disorders; a systematic review by Deepmala and colleagues of clinical trials in neurology and psychiatry shows initial favourable evidence in a range of such disorders (Deepmala et al., 2015); see also the systematic review of human and non-human animal studies by Bhatti et al. (2017).
Conclusions: There is strong, and accumulating, evidence implicating ER stress and UPR activation in the pathogenesis and pathophysiology of a wide range of progressive neurodegenerative diseases. Large,well-powered, double-blind, placebo-controlled trials of potential therapeutic agents which target related pathways need to be carried out in such diseases.
Basant K. Puri*, Gerwyn Morris
Department of Medicine, Imperial College London,
Hammersmith Hospital, London, UK (Puri BK)
IMPACT Strategic Research Centre, School of Medicine,Deakin University, Geelong, Australia (Morris G)
*Correspondence to: Basant K. Puri, MA, PhD, MB, BChir,MSc, MMath, FRCPsych, FRSB, basant.puri@imperial.ac.uk.
orcid: 0000-0001-6101-0139 (Basant K. Puri)
Accepted: 2018-07-16
doi: 10.4103/1673-5374.238614
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open access statement: This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike 4.0 License, which allows others to remix,tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
- 中国神经再生研究(英文版)的其它文章
- Restoration of an injured lower dorsal ascending reticular activating system in a patient with intraventricular hemorrhage
- Taurine protects against retinal and optic nerve damage induced by endothelin-1 in rats via antioxidant effects
- SIRT1 facilitates amyloid beta peptide degradation by upregulating lysosome number in primary astrocytes
- Cognitive deficits and Alzheimer-like neuropathological impairments during adolescence in a rat model of type 2 diabetes mellitus
- Enriched environment elevates expression of growth associated protein-43 in the substantia nigra of SAMP8 mice
- Achyranthes bidentata polypeptide protects dopaminergic neurons from apoptosis induced by rotenone and 6-hydroxydopamine