
Cognitive deficits and Alzheimer-like neuropathological impairments during adolescence in a rat model of type 2 diabetes mellitus
2018-09-11 07:27LiJinYiPeiLiQiongFengLiRenFangWangGuoJiaBoLiWang
中国神经再生研究(英文版) 2018年11期
Li Jin , Yi-Pei Li , Qiong Feng , Li Ren Fang Wang Guo-Jia Bo Li Wang
1 Department of Pathophysiology, Henan Medical College, Zhengzhou, Henan Province, China
2 Henan Key Laboratory of Degenerative Brain Disease, Henan Medical College, Zhengzhou, Henan Province, China
3 Department of Pathophysiology, School of Basic Medicine and the Collaborative Innovation Center for Brain Science, Key Laboratory of Ministry of Education of China for Neurological Disorders, Tongji Medical College, Huazhong University of Science and Technology, Wuhan,Hubei Province, Chinsa
4 Department of Pathology, Wuhan Children’s Hospital, Wuhan, Hubei Province, China
Abstract Numerous studies have shown that many patients who suffer from type 2 diabetes mellitus exhibit cognitive dysfunction and neuronal synaptic impairments. Therefore, growing evidence suggests that type 2 diabetes mellitus has a close relationship with occurrence and progression of neurodegeneration and neural impairment in Alzheimer’s disease. However, the relationship between metabolic disorders caused by type 2 diabetes mellitus and neurodegeneration and neural impairments in Alzheimer’s disease is still not fully determined. Thus, in this study, we replicated a type 2 diabetic animal model by subcutaneous injection of newborn Sprague-Dawley rats with monosodium glutamate during the neonatal period. At 3 months old, the Barnes maze assay was performed to evaluate spatial memory function. Microelectrodes were used to measure electrophysiological function in the hippocampal CA1 region. Western blot assay was used to determine expression levels of glutamate ionotropic receptor NMDA type subunit 2A (GluN2A) and GluN2B in the hippocampus.Enzyme-linked immunosorbent assay was used to determine levels of interleukin-1β, tumor necrosis factor α, and interleukin-6 in the hippocampus and cerebral cortex, as well as hippocampal amyloid beta (Aβ)1–40 and Aβ1–42 levels. Our results showed that in the rat model of type 2 diabetes mellitus caused by monosodium glutamate exposure during the neonatal period, latency was prolonged and the number of errors increased in the Barnes maze. Further, latency was increased and time in the escape platform quadrant shortened. Number of times crossing the platform was also reduced in the Morris water maze. After high frequency stimulation of the hippocampus, synaptic transmission was inhibited, expression of GluN2A and GluN2B were decreased in the hippocampus, expression of interleukin 1β, interleukin 6,and tumor necrosis factor α was increased in the hippocampus and cortex, and levels of Aβ1–40 and Aβ1–42 were increased in the hippocampus. These findings confirm that type 2 diabetes mellitus induced by neonatal monosodium glutamate exposure results in Alzheimer-like neuropathological changes and further causes cognitive deficits and neurodegeneration in young adulthood.
Key Words: nerve regeneration; type 2 diabetes mellitus; Alzheimer’s disease; monosodium glutamate; neonatal period; cognitive deficits;hyperglycemia; hyperinsulinemia; insulin resistance; neural regeneration
Introduction
Type 2 diabetes mellitus (T2DM) is one of the most common glycometabolic disorders (Chen et al., 2011) and is characterized by insulin resistance that reflects a relative insulin deficiency (Kahn, 2003; Muoio and Newgard, 2008).Like Alzheimer’s disease (AD), T2DM can also cause a cognitive disorder and impaired neuronal synaptic structure and function (Luchsinger, 2012). Metabolic disorders(including hyperglycemia, hyperinsulinemia, and insulin resistance caused by T2DM) are considered to be independent risk factors for developing AD (Haan, 2006; Vagelatos and Eslick, 2013). Moreover, epidemiological surveys have found that 70–80% of patients with AD have T2DM or show blood glucose or insulin level abnormalities (Janson et al., 2004; Mwamburi and Qiu, 2016). Thus, an increasing number of studies have considered that metabolic disorders caused by T2DM have a close relationship with the occurrence and progression of neurodegeneration and neural impairment in AD (Barbagallo and Dominguez, 2014; Sridhar et al., 2015).
AD is the most common neurodegenerative disorder in the elderly and is characterized clinically by progressive deterioration of memory and cognitive dysfunction (Scheltens et al., 2016) caused by neural loss and degeneration, extracellular precipitation of amyloid beta (Aβ), and intracellular tau hyperphosphorylation (Nisbet et al., 2015; Lewis and Dickson, 2016; Selkoe and Hardy, 2016). Diabetes mellitus shares many pathophysiological similarities with AD (Bahrmann et al., 2012; Barbagallo and Dominguez, 2014). These similarities include degenerative processes, Aβ aggregation,mitochondrial dysfunction, dyslipidemia, protein hyperphosphorylation, oxidative stress response, and inflammatory response (Li and Hölscher, 2007; Blázquez et al.,2014; De Felice and Ferreira, 2014; Mushtaq et al., 2014),which can lead to neural impairment and degeneration.Even if hyperglycemia cannot cause diabetic symptoms in the early stage of T2DM, it can lead to central nervous system impairment and aggravate cognitive dysfunction(Cox et al., 2005; Biessels et al., 2014; Hamed, 2017). Disrupted insulin metabolism is also associated with cognitive dysfunction and synaptic impairment (Gispen and Biessels,2000; Zhao et al., 2004; de la Monte and Wands, 2005; Grillo et al., 2015). Meanwhile, several studies have found that abnormal insulin levels and disruption of insulin receptor signal transduction pathways can cause reduced Aβ degradation, increased Aβ levels (Gasparini et al., 2001; Farris et al., 2003; Miller et al., 2003), and accelerated tau hyperphosphorylation via activation of glycogen synthase kinase 3 beta (Schubert et al., 2004; Planel et al., 2007; Liu et al.,2011). In addition, hyperinsulinemia has been found in patients with AD and an animal model of AD (Hiltunen et al.,2012; Barron et al., 2013; Mwamburi and Qiu, 2016). Many studies have also observed insulin insensitivity and reduced insulin receptor activity in the brain of AD patients (Bosco et al., 2011; Schioth et al., 2012; Blázquez et al., 2014). Because of disordered insulin and insulin signaling pathways in the AD brain, AD was even thought to be type 3 diabetes in a study conducted by Steen et al. (2005). Consequently,further understanding of the relationship between metabolic disorders caused by diabetes mellitus and neuropathy in AD has positive implications for research and clinical treatment of AD and diabetes.
Monosodium glutamate (MSG)-treated rodents are used as an animal model of T2DM for the study of glycometabolic diseases (Ribeiro et al., 1997; Iwase et al., 1998). Previous studies have reported that MSG exposure can cause cognitive dysfunction in an animal model (Sasaki-Hamada et al., 2015; Madhavadas et al., 2016; Franco et al., 2017).However, none of these studies have examined the occurrence and progression of neurodegeneration and neural impairments in AD. Indeed, we are interested in whether metabolic disorders induced by MSG exposure can cause cognitive deficits and Alzheimer-like neuropathological impairments and neurodegeneration. In particular, we are interested in the effects of neonatal MSG exposure on cognitive deficits and neural function and in the underlying links between metabolic disorders caused by T2DM and AD neuropathy.
In this study, we investigated the effects of T2DM caused by MSG on cognitive capacity and synaptic function of Sprague-Dawley (SD) rats. Further, we investigated the links between metabolic disorders caused by T2DM and neuropathy of AD using behavioral, electrophysiology, molecular biology, and enzyme-linked-immunosorbent assay(ELISA) tests in SD rat models of T2DM.
Materials and Methods
Animals
Ten 18-day-old pregnant SD rats were obtained from the Laboratory Animal Center of Henan Province of China (license number: SCXK (Yu) 2015-0004). In total, 20 neonatal male SD rats were used in this study. Neonatal rats were selected from 10 different litters. Based on treatments with or without MSG (Sigma-Aldrich, St. Louis, MO, USA), all pups were randomly divided into two groups. Experimental pups were administered 50% water-soluble MSG by subcutaneous injection at a dosage of 4 mg/g body weight at postnatal days 1, 3, 5, 7, and 9. Control pups were treated with the same volume of normal saline (0.008 mL/g).All animals were housed in polypropylene cages (4–5 rats per cage), and maintained in a light cycle-controlled (12-hour light/dark cycle) environment with 23 ± 1°C and 50± 10% relative humidity. All pups were fed by their mother until weaning at 28 days and were then allowed free access to regular diet and water. All animal experiments conformed to the ‘Policies on the Use of Animals and Humans in Neuroscience Research’ revised and published by the Society for Neuroscience in 1995, and the animal study was approved by the Academic Review Board of Henan Medical College of China (approval number: 170301001).All rats were sacrificed at the end of behavioral tests for further experiments.
Blood sample assays
Blood samples of all rats were collected from the tail vein after fasting for 12 hours at 3 months old. Levels of fasting blood glucose (FBG) were measured using a glucose test meter (LifeScan, Inc., New Brunswick, NJ, USA). Levels of fasting insulin (FINS) were determined using a radioimmunoassay kit (Chinese Institute of Atomic Energy, Beijing,China). The insulin sensitivity index (ISI) was calculated by: Ln [1 / (FBG × FINS)], with FBG expressed as mM and FINS as mU/L.
Barnes maze assay
All rats were subjected to the Barnes maze assay (Techman, Chengdu, China) to evaluate their spatial memory function at 3 months old. For the Barnes maze assay, rats were trained to find a hole that connected to a black escape box, which was positioned around the circumference of a circular platform. The circular platform was 115 cm diameter and 1.5 cm thick, with 20 evenly spaced 7 cm diameter holes at the edges. It was brightly illuminated by 4 overhead lights as an aversive stimulus. Each trial was recorded by a video camera installed over the platform. Rats were trained for five consecutive days (two trials per day). In each trial,the rat was placed in the center of the platform under a cylindrical black chamber for 10 seconds before the chamber was lifted. When the chamber was lifted, the rat was allowed to locate the target hole and enter the escape box in the allowed time (180 seconds). Each trial ended when the rat had entered the escape box in 180 seconds. When the rat failed to locate the target hole in 180 seconds, it was guided into the escape box and stayed there for 60 seconds. On the sixth day, the escape box was removed for the test. Primary latency to the target hole and the number of errors while exploring during the test were recorded. Between trials, the platform surface and escape box were cleaned with 70%ethanol and water.
Morris water maze assay
Spatial memory function of rats was assessed using the Morris water maze test (Techman) at 3 months old. The Morris water maze assay was performed in a circular pool(60 cm high and 160 cm in diameter) filled with water to a depth of 22 cm. The pool water was opacified using dry milk, and the water temperature maintained between 22 and 24°C. The circular pool was virtually divided into four quadrants. A glass platform was submerged 1.5 cm below the water surface in the third quadrant. During the training session, rats were trained to locate the platform for five continuous days (four times per day). Briefly, rats were manually placed into the water facing the wall of the pool from each of four cardinal start locations and allowed to find the platform for a maximum of 60 seconds per training trial. Each training trial was automatically terminated when the animal climbed onto the platform in 60 seconds and then stayed there for 15 seconds. Rats that failed to find the platform in 60 seconds were manually placed onto the platform and stayed there for 15 seconds. For each rat,the swimming path and time taken to locate the escape platform was recorded in each training trial using the Noldus video tracking system (Ethovision; Noldus Information Technology, Beijing, China). Following training trials, the platform was removed for the probe trial on the sixth day.The percentage time spent in each quadrant and total times crossing the place where the escape platform was located were recorded.
Electrophysiological measure
Rats were deeply anesthetized by intraperitoneal injection with 10% chloral hydrate (0.6 mL/100 g). Brains were removed and coronal slices (300-μm thick) cut using a vibratome (Leica, Wetzlar, Germany) in ice-cold artificial cerebrospinal fluid containing (in mM): NaCl (Sigma-Aldrich)126, KCl 3.0, MgCl21.0, CaCl22.0, NaH2PO41.25, NaHCO326, and glucose 10, saturated with 95% O2, 5% CO2(pH 7.4). Subsequently, slices were transferred to an incubation chamber filled with oxygenated artificial cerebrospinal fluid to recover for 1 hour before testing.
For long-term potentiation (LTP), slices were placed on a chamber with an 8 × 8 microelectrode array in the bottom planar (each 50 μm × 50 μm in size, with an interpolar distance of 450 μm), and submerged in artificial cerebrospinal fluid (4 mL/min) with a nylon silk glued to a platinum ring.Signals were acquired using the MED64 System (Alpha MED Sciences, Panasonic, Osaka, Japan), as described in a previous study (Yin et al., 2016). Field excitatory postsynaptic potentials in the CA1 region were recorded by stimulating Schaffer fibers from CA3. Stimulation intensity was adjusted to evoke field excitatory postsynaptic potential amplitudes that were 40% of maximal size. LTP was induced by applying one train of high-frequency stimulation (100 Hz, 1 second duration at test strength).
Western blot assay
Rats were decapitated under deep anesthesia, and hippocampi quickly dissected from the head on ice and stored at−80°C. Hippocampi were homogenized in buffer containing: NaCl 50 mM, Tris (Sigma-Aldrich) 10 mM, ethylenediamine tetraacetic acid 1 mM, Na3VO40.5 mM, NaF 50 mM, phenylmethyl sulfonylfluoride 1 mM, and a protease-inhibitor cocktail (Sigma-Aldrich; P8340). Afterwards,extracts were centrifuged at 14,000 × g for 10 minutes at 4°C,boiled for 10 minutes in water at 130°C, and then stored at−80°C for western blot assay. For western blot assays, equal quantities of protein samples were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. Next, membranes were incubated with rabbit polyclonal antibody anti-glutamate ionotropic receptor NMDA type subunit 2A (GluN2A,1:1000; Abcam, Cambridge, MA, USA), rabbit polyclonal antibody anti-glutamate ionotropic receptor NMDA type subunit 2B (GluN2B, 1:1000; Abcam), and mouse monoclonal antibody anti-α-tubulin (DM1A, 1:1000; Sigma-Aldrich)at 4°C overnight after blocking with 5% (w/v) non-fat milk/Tris-buffered saline with Tween-20 for 1 hour. Finally, blots were probed with goat anti-mouse (for DM1A, 1:10,000) or goat anti-rabbit (for GluN2A and GluN2B, 1:10,000) IgG conjugated to IRDye (800CW, Licor Biosciences, Lincoln,NE, USA) for 1 hour at room temperature, and then analyzed using the Odyssey infrared imaging system (Licor Biosciences).
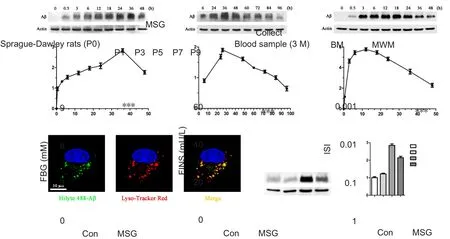
Figure 1 Effect of MSG exposure during the neonatal period on levels of FBG, FINS, and ISI value in 3-month-old rats.
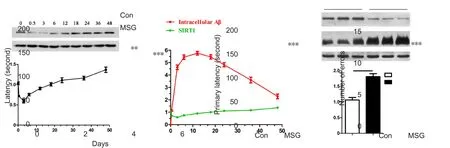
Figure 2 Effect of MSG exposure during the neonatal period on learning and spatial memory in 3-month-old rats in the Barnes maze.
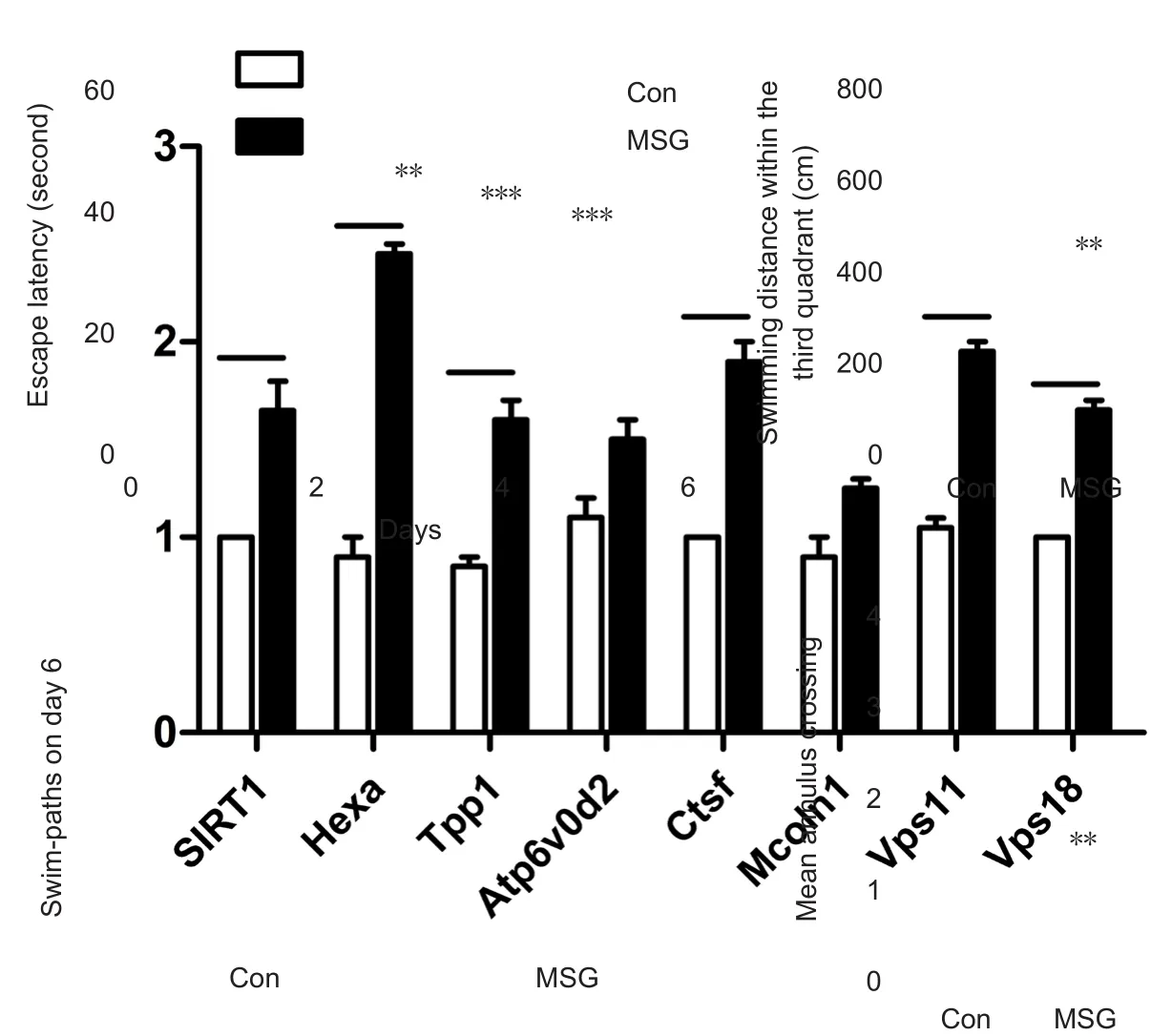
Figure 3 Effect of MSG exposure during the neonatal period on learning and spatial memory in 3-month-old rats using the Morris water maze.
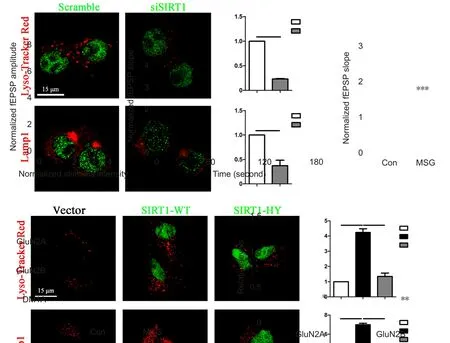
Figure 4 Effect of MSG exposure during the neonatal period on neuronal synaptic plasticity of the hippocampus of 3-month-old rats.
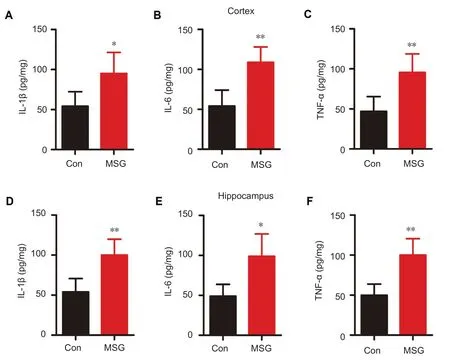
Figure 5 Effect of MSG exposure during the neonatal period on expression of in flammatory cytokines in the hippocampus and cortex of 3-month-old rats.

Figure 6 Effect of MSG exposure during the neonatal period on hippocampal concentration of Aβ in 3-month-old rats.
ELISA
Concentrations of interleukin-1 beta (IL-1β), tumor necrotic factor-alpha (TNF-α), and interleukin-6 (IL-6) in the hippocampus and cortex were determined using rat ELISA kits (Diaclone, London, UK), according to the manufacturer’s protocol. Hippocampal concentrations of Aβ1–40and Aβ1–42were detected using ELISA kits (Biosource International, Inc., Camarillo, CA, USA), in accordance with the manufacturer’s protocol. Results were expressed as pg/mg of protein.
Statistical analysis
Data were expressed as the mean ± SD. Comparison among two groups was performed by independent-sample t-test using SPSS 18.0 software (IBM, Armork, NY, USA). P-values of < 0.05 were accepted as statistically significant.
Results
MSG exposure during the neonatal period increased FBG and FINS levels and reduced ISI value in 3-month-old rats
To replicate the T2DM animal model, newborn SD rats were administered MSG by subcutaneous injection at postnatal days 1, 3, 5, 7, and 9. Blood samples were collected to examine FBG and FINS levels in the rats at 3 months old(Figure 1A). MSG exposure during the neonatal period significantly increased levels of FBG (Figure 1B; P < 0.001)and FINS (Figure 1C; P < 0.001) in 3-month-old rats compared with age-matched rats from the control group. ISI value was also reduced in 3-month-old MSG-treated rats compared with age-matched rats from the control group(Figure 1D; P < 0.001). Altogether, this indicates that the type 2 diabetic animal model was successfully replicated in our study.
T2DM caused by MSG exposure during the neonatal period led to cognitive deficits in 3-month-old rats
Cognitive dysfunction and memory loss are principal clinical manifestations in patients with AD (Scheltens et al.,2016). To investigate the effect of T2DM caused by MSG exposure during the neonatal period on cognitive ability, the Barnes maze assay was first performed to examine cognitive ability in rats at 3 months old. Our results show obvious cognition and memory deficits in MSG-treated rats compared with age-matched rats from the control group. Latency (taken time to enter the escape chamber)was significantly increased from the fourth day of training in MSG-treated rats compared with age-matched rats of the control group (Figure 2A; Pday4= 0.003, Pday5< 0.001). MSG exposure also decreased learning and memory abilities of MSG-exposed rats, as shown by significantly increased primary latency to enter the escape chamber and number of errors during the test (Figure 2B and C; Pprimarylatency< 0.001,Pnumbersoferrors< 0.001).
To further examine the effect of T2DM caused by MSG exposure during the neonatal period on cognitive ability,the Morris water maze assay was also performed. Similarly, our results also showed that MSG exposure decreased cognitive ability of MSG-treated rats in the Morris water maze assay. Latency to reach the platform was significantly increased from the third day of training in MSG-exposed rats compared with age-matched rats of the control group(Figure 3A; Pday3= 0.02, Pday4< 0.001, Pday5< 0.001). Further, on day 6 of the testing session, MSG-treated rats were less likely to remain in the quadrant where the escape platform had been located (Figure 3B and C; P = 0.005) and also exhibited a significantly decreased number of platform crossings compared with the control group (Figure 3D; P =0.001).
T2DM caused by MSG exposure during the neonatal period impaired hippocampal neuronal synaptic plasticity in 3-month-old rats
Neuronal synaptic plasticity is critical for learning and memory function (Lamprecht and LeDoux, 2004). Progression of AD can cause impairment of neuronal synaptic plasticity, which correlate with cognitive deficits caused by AD (Jacobsen et al., 2006; Shankar et al., 2008). In the next part of our study, we investigated LTP in the hippocampus,a neuroelectrophysiological measure related to learning and memory function (Lynch, 2004), by recording field excitatory postsynaptic potentials after tetanic stimulation.Extracellular field excitatory postsynaptic potentials were recorded in the CA1 stratum radiatum of acute slices in response to stimulation of Schaffer collateral input from CA3. Our results found that T2DM caused by MSG exposure during the neonatal period suppressed basal synaptic transmission, as shown by input-output curves (Figure 4A;P3stimulusintensity= 0.014, P3.5stimulusintensity= 0.009) and attenuated slope of field excitatory postsynaptic potentials after high-frequency stimulation in the hippocampus (Figure 4B and C; P < 0.001) compared with age-matched rats from the control group.
To investigate the underlying molecular mechanism of LTP impairment, western blot assay was performed to examine expression changes of synaptic plasticity-related proteins in the hippocampus of rats that underwent electrophysiological testing. Our results show that T2DM caused by MSG exposure during the neonatal period decreased expression levels of synaptic plasticity-related proteins. Specifically, we found significantly reduced expression levels of GluN2A and GluN2B in the hippocampus compared with age-matched rats from control group (Figure 4D and E;PGluN2A= 0.033, PGluN2B= 0.003).
T2DM caused by MSG exposure during the neonatal period resulted in high expression of in flammatory cytokines in the hippocampus and cortex of 3-month-old rats
Inflammatory responses and activation of inflammatory signaling pathways are common phenomena in the AD brain (Heneka and O’Banion, 2007). Expression levels of inflammatory cytokines in the hippocampus and cortex were detected by ELISA. Our results show that IL-1β,IL-6, and TNF-α expression levels were significantly increased in the hippocampus and cortex of MSG-exposed rats compared with age-matched rats from the control group (Figure 5; cortex: PIL-1β= 0.024, PIL-6= 0.002, PTNF-α= 0.007; Hip: PIL-1β= 0.004, PIL-6= 0.012, PTNF-α= 0.003).This indicates that T2DM caused by MSG exposure leads to an in flammatory response in 3-month-old rats.
T2DM caused by MSG exposure during the neonatal period increased Aβ1–40 and Aβ1–42 concentration in the hippocampus of 3-month-old rats
Extracellular deposition of Aβ in the brain of patients with AD is a main neuropathological characteristic of AD(Selkoe, 1996). Thus, concentration of Aβ1–40and Aβ1–42in the hippocampus was measured using ELISA kits. We found that Aβ1–40and Aβ1–42concentration was significantly increased in the hippocampus of MSG-exposed rats compared with age-matched rats from the control group (Figure 6; PAβ1–40= 0.013, PAβ1–42= 0.012). This indicates that T2DM caused by MSG exposure leads to Alzheimer-like neuropathological impairments in 3-month-old rats.
Discussion
Diabetes mellitus and AD are both age-related chronic degenerative diseases, which pose a serious hazard to the health of aged people (Pasquier et al., 2006). Diabetes mellitus can cause impairment of the central nervous system and severe deterioration of cognitive capacity (Gispen and Biessels, 2000; Kodl and Seaquist, 2008; McCrimmon et al.,2012). An increasing number of investigators have considered that metabolic disorders caused by diabetes mellitus have a close relationship with progression of AD (Steen et al., 2005; Sridhar et al., 2015; Kandimalla et al., 2017). However, the underlying mechanism and relationship between metabolic disorders caused by T2DM and neuropathy of AD are still not fully elucidated. Our results here show that neonatal exposure to MSG can lead to overexpression of Aβ and an inflammatory response in young adulthood (3 months), which further causes cognitive dysfunction and neurodegeneration (impairment of synaptic function and loss of synaptic molecules). This indicates that metabolic disorders caused by T2DM can lead to neurodegeneration and Alzheimer-like neuropathological changes in young adulthood, which ultimately contributes to further understanding of the effect of metabolic disorders caused by T2DM on occurrence and progression of AD. Furthermore,occurrence and progression of cognitive dysfunction and neurodegeneration show a strong relationship with overexpression of Aβ and inflammatory response induced by MSG exposure, which may reflect the underlying mechanism of Alzheimer-like symptoms and neuropathy caused by T2DM.
As a salt of glutamic acid, MSG is used as a flavoring agent in Asian countries (Jinap and Hajeb, 2010). In the 1970s, a previous study involving the effect of subcutaneous MSG injection during the neonatal period of rats revealed that early MSG exposure has detrimental effects on rat growth, reproductive and brain function, as well as impaired glucose tolerance (Lengvári, 1977). Further, many studies in the late 1990s also found that MSG exposure can lead to development of many common clinical pathological symptoms of T2DM, such as insulin resistance,hyperinsulinemia, glucose intolerance, and hyperglycemia in experimental animals (Hirata et al., 1997; Ribeiro et al., 1997; Iwase et al., 1998). Furthermore, several studies have found that complications of diabetes mellitus (such as atherosclerosis, hypercholesterolemia, central obesity and hypertension) can be induced by early MSG exposure(Iwase et al., 1998; Nagata et al., 2006; Morrison et al.,2008). Therefore, in recent years, an increasing number of studies have served as MSG-treated animals as a model of T2DM (Islam and Wilson, 2012). MSG was also used to replicate animal models of T2DM in our study. Moreover,we found that MSG exposure during the neonatal period increased FBG and FINS levels and reduced ISI value. ISI reflects insulin sensitivity on regulation of blood glucose.Reduction of ISI in experimental animals signifies insulin resistance, which indicates that the type 2 diabetic animal model was successfully replicated in this study. Neuronal synaptic structure and function in the hippocampus are strongly associated with learning and memory ability (Bliss and Collingridge, 1993; Santin et al., 2000). Impairment in structure and function of neuronal synapses is a major characteristic of neuronal structural pathology in the AD brain and is considered to lead to cognitive deficits in patients with AD (Arendt, 2009; Metaxas and Kempf,2016; Zhang et al., 2016). In particular, much evidence has demonstrated that AD progression is able to cause impairment of neuronal synaptic plasticity in the hippocampus(Chapman et al., 1999; Walsh et al., 2002; Oddo et al.,2003; Lacor et al., 2004). The results of our study show that MSG exposure during the neonatal period results in cognitive deficits and impaired LTP of hippocampal neurons in 3-month-old rats. LTP is a major reflection of synaptic plasticity and is regarded as a principal cellular mechanism involved in learning activity and memory formation. Inhibition of LTP in this study suggests that T2DM induced by MSG exposure causes impaired synaptic plasticity function in the hippocampus, which may be an underlying link between structural pathology and AD.
Synaptic-related proteins are the molecular biological basis of neuronal synaptic function, with changes in these proteins directly impacting on learning and memory func-tion (Izquierdo and Medina, 1997; Riedel et al., 2003). Accordingly, we next performed western blot assays to further investigate the underlying molecular mechanisms of LTP impairment. Our results show that MSG exposure during the neonatal period decreases expression levels of synaptic plasticity-related proteins, including GluN2A and GluN2B.GluN2A and GluN2B play critical roles in excitatory synaptic transmission and synaptic plasticity in the central nervous system (Malenka, 1994; Yashiro and Philpot, 2008).LTP requires GluN receptor activation, and the subsequent cascade of events is triggered by Ca2+in flux (Malenka and Bear, 2004). Reduction of GluN2A and GluN2B expression is associated with LTP inhibition (Lau and Zukin, 2007).These molecular assay results are consistent with our electrophysiological analysis.
There are complex links between diabetes mellitus and AD (Yang and Song, 2013; Ninomiya, 2014). The in flammatory response and inflammatory signaling pathway are among the major pathogenic links between diabetes mellitus and AD (Jones et al., 2009; Kamal et al., 2014). Many studies have demonstrated that progression of diabetes mellitus and AD both cause an in flammatory response and trigger high expression of in flammatory cytokines (Granic et al., 2009). Furthermore, the inflammatory response is also regarded as a major pathological process in AD progression (Akiyama et al., 2000). We found that MSG exposure during the neonatal period leads to high expression of in flammatory cytokines in the hippocampus and cortex,which indicates that T2DM induced by MSG exposure can cause an in flammatory response in 3-month-old Sprague–Dawley rats. The in flammatory response and activation of the in flammatory signaling pathway play an important role in AD progression (Heneka and O’Banion, 2007). Much evidence has revealed that the in flammatory response and inflammatory signaling pathway activation are tightly correlated with generation and deposition of Aβ, which is a crucial step in the cascade process of Aβ (Selkoe, 1996;Heneka and O’Banion, 2007). Consequently, our results here suggest that the in flammatory response and in flammatory signaling pathway activation may reflect an underlying link of disease progression between T2DM and AD.
Hippocampal accumulation of amyloid plaques chiefly formed by Aβ deposition is a principal pathological characteristic of AD (Selkoe and Hardy, 2016) and tightly correlates with cognitive dysfunction and neuronal synaptic impairment. Our data show that MSG exposure during the neonatal period significantly increases hippocampal concentration of Aβ1–40and Aβ1–42compared with age-matched rats from the control group. Previous studies have reported that hyperinsulinemia and dysfunction in the insulin receptor signal transduction pathway contribute to reduced Aβ degradation and production of Aβ (Carlsson, 2010).Our data further indicate that T2DM causes Alzheimer-like neuropathological changes, which is a main reason for AD-like neurodegeneration caused by T2DM. Intracellular tau hyperphosphorylation is another main neuropathological characteristic of AD. However, we did not investigate the effect of MSG exposure during the neonatal period on tau phosphorylation levels in this study, and this needs further investigation.
In conclusion, T2DM induced by MSG exposure during the neonatal period leads to Aβ overexpression and an in flammatory response. Ultimately, this leads to cognitive deficits and neurodegeneration, including impairment of synaptic plasticity function and loss of synaptic molecules in young adulthood. Our study provides experimental data for a connection between T2DM and AD.
Acknowledgments: We specially thank Professor Gong-Ping Liu from Huazhong University of Science and Technology of China (Key Laboratory of Ministry of Education of China for Neurological Disorders, Tongji Medical College, China) for discussion of the data, and his revision and proofreading of the manuscript. The same gratitude goes to Mr. Guo-Yong Li and Miss Sha Liu, two current students at Huazhong University of Science and Technology, Wuhan, China
Author contributions: LW and LJ designed this study. LJ and YPL prepared all figures and wrote the manuscript. YPL and LR established experimental animal models and conducted the blood sample assays. YPL, LR and GJB performed the behavioral tests. QF and LJ conducted data analysis. LJ performed the western blot assay. YPL conducted the ELISA assays. All authors approved the final version of the paper.
Conflicts of interest: All authors declare that no potential or actual conflicts of interest including any financial or personal relationships with other people or research organizations within two years of beginning this work submitted that could inappropriately in fluence (bias)their work.
Financial support: This study was principally supported by the Initial Funding of PhD Research from Henan Medical College of China,No. 1001/0106; and in parts by the Science and Technology Project of Henan Province of China, No. 172102310105. All authors declare that financial support does not affect the opinion of the article and the objective statistical analysis and report of the research results in this study.
Institutional review board statement: The study was approved by the Academic Review Board of Henan Medical College of China (approval number: 170301001).
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open access statement: This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer: Aysegul Yildiz-Unal, Mugla Sitki Kocman University, Turkey.
Additional file: Open peer review report 1.
- 中国神经再生研究(英文版)的其它文章
- The unfolded protein response signaling and retinal Müller cell metabolism
- Sequencing of high-efficacy disease-modifying therapies in multiple sclerosis: perspectives and approaches
- Targeting prion-like protein spreading in neurodegenerative diseases
- Cadmium-induced neurotoxicity: still much ado
- Analysis of the traf ficking system in blood-brain barrier models by high content screening microscopy
- Retinal remodeling following photoreceptor degeneration causes retinal ganglion cell death