
The unfolded protein response signaling and retinal Müller cell metabolism
2018-09-11 07:26:48KristenKellyJoshuaWangSarahZhang
中国神经再生研究(英文版) 2018年11期
Kristen Kelly, Joshua J. Wang, , Sarah X. Zhang, ,
1 Department of Ophthalmology and Neuroscience Program, Ross Eye Institute, University at Buffalo, State University of New York, Buffalo, NY, USA
2 SUNY Eye Institute, State University of New York, Buffalo, NY, USA
Abstract The retina is one of the most energy demanding tissues in the body. Like most neurons in the central nervous system, retinal neurons consume high amounts of adenosine-5′-triphosphate (ATP) to generate visual signal and transmit the information to the brain. Disruptions in retinal metabolism can cause neuronal dysfunction and degeneration resulting in severe visual impairment and even blindness. The homeostasis of retinal metabolism is tightly controlled by multiple signaling pathways, such as the unfolded protein response (UPR), and the close interactions between retinal neurons and other retinal cell types including vascular cells and Müller glia. The UPR is a highly conserved adaptive cellular response and can be triggered by many physiological stressors and pathophysiological conditions. Activation of the UPR leads to changes in glycolytic rate, ATP production, de novo serine synthesis, and the hexosamine biosynthetic pathway, which are considered critical components of Müller glia metabolism and provide metabolic support to surrounding neurons. When these pathways are disrupted, neurodegeneration occurs rapidly. In this review, we summarize recent advance in studies of the UPR in Müller glia and highlight the potential role of the UPR in retinal degeneration through regulation of Müller glia metabolism.
Key Words: unfolded protein response; retina; Müller glia; metabolism; neurodegeneration; X-box binding protein 1; glycolysis; glucose transporter
Introduction
The neural retina is made up of cells that can be classified into two major types: neurons and glial cells and each of these cell types consists of multiple sub-populations. Retinal neurons include bipolar cells, amacrine cells, horizontal cells, ganglion cells, and photoreceptors. Rod and cone photoreceptors hyperpolarize in response to dim and bright light, respectively (Masland, 2012). Horizontal cells send inhibitory inputs to photoreceptors to help the cells respond to changes in the brightness of light. Bipolar cells along with amacrine cells help transmit the photoreceptor signal to retinal ganglion cells (RGCs), whose axons exit the retina through the optic disc and form the optic nerve to transmit the information to the brain. Three different glial cell types are present in the retina: astrocytes, microglia, and Müller glia. Müller glia and astrocytes are characterized as macroglial cells because of their ability to support surrounding neurons and maintain the blood-retina barrier (BRB) (Kolb,1995). Müller glia cell bodies are located in the inner nuclear layer, but they have long processes that extend in both directions and allow them to interact with retinal neurons and vascular cells. Their interactions with microglia are thought to increase upon disruptions to retinal homeostasis, wherein microglia release signaling molecules to trigger functional responses in Müller glia (Wang and Wong, 2014).
Retinal neurons, particularly photoreceptors, require a massive amount of energy to function. In general, retinal neurons rely on mitochondrial oxidative phosphorylation for energy, whereas glial cells rely on both oxidative phosphorylation and glycolysis (Winkler et al., 2000). Under metabolically stressful conditions, such as low glucose or anoxia, Müller glia can increase their rate of glycolysis in order to obtain ATP (Toft-Kehler et al., 2018). Knowing the limit to which Müller glia are capable of adapting to metabolic stress, and elucidating how the cells respond to the stress, is important for understanding their function in supporting neuronal cells and their clinical significance in preventing retinal degenerative disorders. Several major signaling pathways have been identified to regulate metabolism and Müller glia stress responses, among which is the unfolded protein response (UPR) (Zhong et al., 2012; Liu et al., 2013; Wang et al., 2014; Gao et al., 2015; Chen et al., 2016a). In this review,we summarize the studies on potential implications of the UPR in regulation of Müller glia stress response and glucose metabolism. Because retinal neurons in humans do not regenerate, a comprehensive understanding of the regulatory mechanisms for Müller glia and retinal metabolism is essential for developing optimal approaches to alleviate metabolic stress, maintain neuronal function, reduce cell death, and ultimately preserve vision in retinal degenerative disorders.
Retinal Metabolism
In general, neural tissues have very high energy demand to maintain their routine activities. For example, the brain consumes 20% of the body’s oxygen and 25% of the body’s glucose stores. This amount of energy consumption is astonishing relative to the small percentage of body mass(2%) it accounts for (Rolfe and Brown, 1997; Wong-Riley,2010). The high metabolic needs is believed to be due primarily to the ion transport in neurons, which actively pump ions against their concentration gradients to maintain the polarization of neuronal membrane (Wong-Riley, 2010).Compared to the brain, the retina is even more energy demanding. Membrane polarization of retinal photoreceptors is essential for the cells to maintain the dark current and respond to changes in light levels. Photoreceptors, along with all other retinal neurons, primarily use oxidative phosphorylation for energy generation due to the higher adenosine triphosphate (ATP) yield compared to glycolysis. In contrast, glial cells rely on glycolysis and have the capacity for oxidative phosphorylation if needed (Winkler et al., 2000).The significant difference in cellular activities and metabolic needs of retinal neurons and glial cells renders retinal metabolism highly compartmentalized (Hurley et al., 2015). For example, RGCs, whose axons are unmyelinated within the retina and the optic nerve head, require continuous blood and energy supply to maintain their viability and function;part of this high energy demand is met by obtaining metabolic substrates including pyruvate and lactate from the surrounding Müller cells (Rueda et al., 2016; Toft-Kehler et al.,2017, 2018). In addition, certain retinal cell types are found more vulnerable to metabolic stress induced cell death than others (Rueda et al., 2016). For instance, under hypoxic conditions, retinal neurons undergo apoptosis much quicker than glial cells (Xin et al., 2013). Different types of retinal neurons also display distinct survivability (Ait-Ali et al.,2015). In a swine model of photoreceptor degeneration,when glycolysis is inhibited by iodoacetic acid (IAA), retinal rod cells die before the cones (Wang et al., 2011).
Metabolic compartmentalization in the retina is also attributable to the anatomical structure of the retina. The neurosensory retina receives blood supply from two sources: the central retinal artery and the posterior ciliary arteries; both originate from the ophthalmic artery. Specifically, the inner two-thirds of the neural retina is nourished by the central retinal artery. The outer retina, primarily consisting of retinal photoreceptor cells, is supplied by the choroidal blood vessels (Pournaras et al., 2008). The outer plexiform layer receives blood supply from both the retinal and choroidal vascular systems. Due to the differences in blood supply, the inner retina is relatively more hypoxic than the outer retina(Yu and Cringle, 2001). This leads to a significant need for Müller glia to provide additional metabolic support to the inner retinal neurons. Despite the distinct blood supply systems, both outer and inner retinal neurons are protected by the BRB, which is formed by the tight junctions between vascular endothelial cells or retinal pigmented epithelium(RPE) cells in the inner or outer retina, respectively. Glucose derived from the blood supply reaches retina cells by crossing the BRB via glucose transporters expressed on the cells of the inner and outer BRB. Glucose transporter 1 (GLUT1/SLC2A1) is the primary glucose transporter expressed across these membranes and is responsible for maintaining the basal level of glucose in the retina (Kumagai et al., 1994; Chen et al., 2016a). Expression of GLUT1 changes in response to glucose levels: increasing when glucose availability is low and decreasing when glucose availability is high. Importantly, Müller glia play a critical role in mediating retinal neurovascular coupling. Upon activation, retinal neurons release ATP, which stimulates purinergic receptor on glial cells. In turn, glial cells produce vasoactive metabolites resulting in vasodilation and vasoconstriction of retinal blood vessels. In disease conditions such as diabetic retinopathy, dysfunction and loss of Müller cells leads to impaired neurovascular coupling contributing to retinal ischemia and neuronal injury(Metea and Newman, 2006; Newman, 2015).
Anatomical Features and Functions of Müller Glia
The structure of the retina itself provides clear support for the critical role of Müller glia in retinal health. Müller glia span the entire width of the retina and thereby provide important structural support to other cell populations(Kolb, 1995). Additionally, their size allows them to project processes that interact with all types of retinal neurons in each of the retinal layers (Figure 1). Müller glia are oriented radially in the retina such that their end feet form the internal limiting membrane that separates the retina from the vitreous, their cell bodies sit in the inner nuclear layer,and their apical processes extend out posteriorly to form the outer limiting membrane, which divides the inner and outer segments of photoreceptors (Germer et al., 1998; Winkler et al., 2000; Roesch et al., 2008; Toft-Kehler et al., 2018). The endfoot processes of Müller glia also directly contact the large blood vessels in the super ficial retinal layer and wrap around capillaries in the deep retina. The position of Müller cells in the retina makes other cell populations particularly susceptible to changes in their functionality.
Müller glia are the primary glial cells of the retina and are crucial in providing support to inner retinal neurons. They help maintain the extracellular environment in several ways.Müller glia process surround many synaptic connections of the retina, allowing them to closely monitor neurotransmitter release, reuptake, and degradation- primarily of excitatory neurotransmitter glutamate (Newman and Reichenbach,1996; Thoreson and Witkovsky, 1999). Neurotransmission of visual signals relies on glutamate release; pathology of the inner retina is often characterized by increases in glutamate levels (Rauen, 2000; Rauen and Wiessner, 2000; Bringmann et al., 2009). They also regulate potassium uptake and siphoning to ensure that membrane concentration gradients of retinal neurons are maintained. Additionally, they are capable of buffering retinal pH to prevent acidification of the retina, which can lead to disturbances in neurotransmission.
In addition to their capacity to regulate retinal metabolites, the proximity of Müller glia to inner retinal neurons,particularly RGCs, supports the idea of metabolic coupling of glia and neurons. Müller glia synthesize, store, and degrade glycogen in their mitochondria rich end feet, and shuttle the cellular energy substrates lactate and pyruvate to surrounding neurons (Perezleon et al., 2013; Hurley et al., 2015). It is worth noting that Müller glia in vascularized retinas, such as in humans and mice, contain higher level of glycogen stores and a more evenly distributed mitochondria,due to greater access to oxygen stores than the cells in avascularized retinas (Toft-Kehler et al., 2017, 2018). Müller cells exposed to high glucose or glucose deprivation demonstrate reduced mitochondrial activity, increased mitochondrial fragmentation, and enhanced apoptosis (Toft-Kehler et al.,2016; Tien et al., 2017). This suggests that in diseased states,such as diabetic retinopathy, Müller glia can suffer mitochondrial stress and damage. Mitochondrial defects lead to Müller cell dysfunction and pathologies, which in turn results in glutamate excitotoxicity in the retina and neuronal death, especially of RGCs (Thoreson and Witkovsky, 1999;Rauen and Wiessner, 2000; Vohra et al., 2017).
Another important function of Müller glia is to produce neurotrophic factors and cytokines to maintain the normal function of neuronal and vascular cells. For example, Müller glia secrete several neurotrophic factors including pigment epithelium-derived factor (PEDF), glial cell like derived neurotrophic factor (GDNF), and brain-derived neurotrophic factor (BDNF), which are found to be essential for retinal neuronal survival and function and for maintaining the BRB integrity (Zhang et al., 2006a; Fu et al., 2015). In addition,Müller glia have been shown to play a role in regulation of the inflammatory response in pathological conditions,through an increased release of cytokines including monocyte chemoattractant protein 1 (MCP-1) and vascular endothelial growth factor (VEGF) (Nakazawa et al., 2006, 2007).Under stressed conditions including hypoxia, in flammation,or high glucose conditions, Müller glia secrete increased amounts of tumor necrotic factor α (TNF-α) and VEGF,which promote the in flammatory response, disrupt the BRB,and increase vascular leakage, contributing to the pathogenesis of retinal edema in diabetic retinopathy (Mathews et al.,1997; Zhang et al., 2006b; Wang et al., 2010; Zhong et al.,2012; He et al., 2015).
Metabolic Homeostasis and Glycolysis in Müller Glia
Maintenance of metabolic homeostasis within Müller glia and consequently in the retina by Müller glia is a complex process. In conditions of metabolic stress in the retina,retinal neurons particularly photoreceptors, are the first to undergo apoptosis. The increased survivability of Müller glia is believed attributable to their glycogen stores, reliance on glycolytic respiration, and lower energy demand relative to retinal neurons. Müller glia can function for some time in conditions of glucose deprivation, or hypoxia, but when both oxygen and glucose availability are low Müller glia undergo apoptosis more rapidly (Toft-Kehler et al., 2017).Therefore, it would be important to understand the relationship of oxygen and glucose availability, mitochondrial function, and glycolysis in Müller glia under stressed conditions.Glycolysis is one of the most important cellular energy pathways used by eukaryotes. In glial cells, glycolysis is generally favored over oxidative phosphorylation for cellular energy, allotting more energy substrates to the high energy demanding neurons. The glycolytic pathway consists of ten enzyme catalyzed steps which convert glucose to two molecules of pyruvate, with a net yield of 2 molecules of ATP per glucose molecule oxidized. The glycolytic reactions can be categorized into two classes - reversible and irreversible.Three glycolytic enzymes, pyruvate kinase (PK), phosofructokinase (PFK), and hexokinase (HK) catalyze the irreversible reactions. Additionally, three of the enzyme steps are considered to be regulatory including, step 1 catalyzed by HK, step 2 catalyzed by glucose-6 phosphate isomerase(G6P), and step 7 catalyzed by phosphoglycerate kinase(PGK) (Figure 2).
Several studies have characterized the glycolytic activity in Müller glia (Poitry-Yamate and Tsacopoulos, 1991;Kooragayala et al., 2015; Rueda et al., 2016; Skytt et al., 2016;Vallee et al., 2017; Tchernookova et al., 2018). In general,Müller glia rely primarily on aerobic glycolysis to generate ATP and only derive a small amount of ATP from oxidative phosphorylation despite the much higher ATP yield from this process. This characteristic is also known as the Warburg effect, or the predominance of glycolysis over oxidative phosphorylation, which allows Müller glia to spare oxygen for surrounding neurons in hypoxic conditions (Winkler et al., 2000; Roesch et al., 2008; Hurley et al., 2015). Compared to other retinal cells, Müller glia are more viable and can survive in acute conditions of low glucose, oxygen deprivation, or mitochondria inhibition by utilizing different energy pathways. In low glucose conditions they rely on oxidative phosphorylation more than glycolysis, while under hypoxic conditions or mitochondrial inhibition they would utilize glycolysis as the principal metabolic pathway (Tien et al., 2017). However, in conditions with combined glucose deprivation and mitochondrial inhibition, Müller glia will be unable to survive due to the lack of an alternative pathway to produce ATP. The potent glycolytic capacity renders Müller glia more resistant to retinal ischemia than neurons;yet sustained and severe ischemia will deplete their stored glycogen, which is required for glycolysis, leading to cell death.
There has been a debate on whether Müller glia rely solely on the glycolytic pathway, which consists of 10 enzyme catalyzed reactions, or also utilize the pentose phosphate pathway (PPP)/hexose monophosphate shunt (HMPS) pathway, which runs in parallel to glycolysis (Buse et al., 2002;Buse, 2006; Semba et al., 2014). The PPP/HMPS is an alternative glucose oxidation pathway, which converts glucose 6 phosphate ‘shunted’ from the first reaction of glycolysis to ribose-5-phosphate, NADPH, and erythrose-4-phosphate.It is believed that both pathways likely take place in Müller glia, because the cells express genes and enzymes required for these pathways. Furthermore, the PPP generates NADPH through consumption of organic carbon and NADPH is vital for preventing oxidative stress in highly metabolically active retina tissue. Thus, the partial use of the PPP is plausible.
Another important function of Müller glia is to carry out glutamate-glutamine cycling, which is critical for preventing glutamate excitotoxicity in the retina (Bringmann et al.,2009; Vohra et al., 2017). Glutamate released from RGCsduring neurotransmission is taken up by Müller glia through excitatory amino acid transporters (EAATs) and converted to glutamine through glutamine synthase (GS). Glutamine is then released back to retinal neurons, where it is converted back to glutamate for neuronal signal transduction. In conditions of low glucose, glutamate can be metabolized by glutamate dehydrogenase and enter the tricarboxylic acid (TCA)cycle in Müller glia, which couples neurotransmission activity to metabolism. Similarly, Müller glia can utilize lactate released from retinal neurons to synthesize glutamine; thus the levels of extracellular lactate can be used as a marker of retinal energy homeostasis.
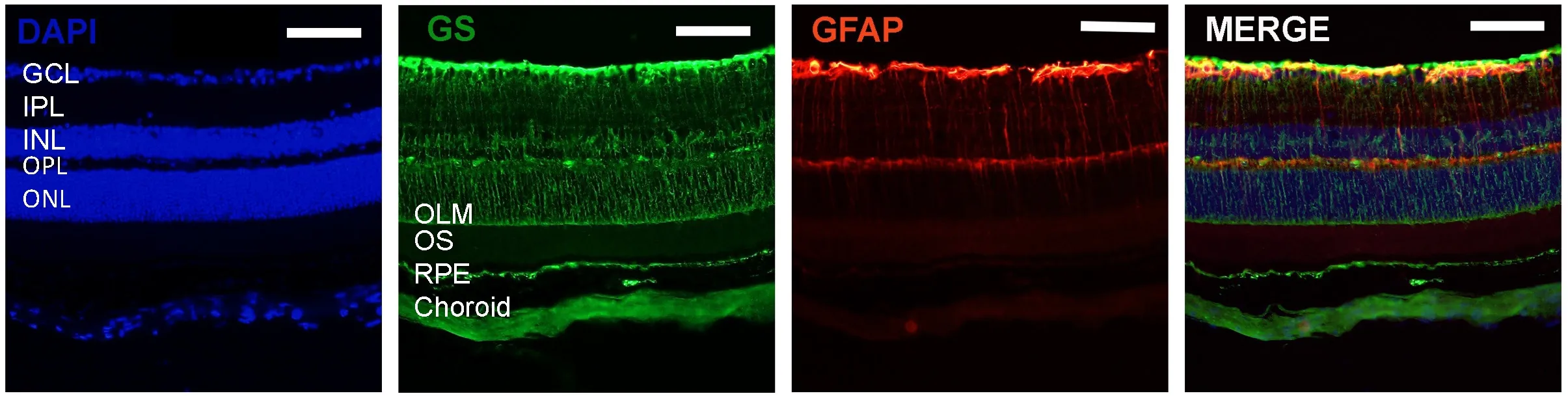
Figure 1 Immunostaining for glutamine synthetase (GS) and glial fibrillary acidic protein (GFAP) in mouse retinas.
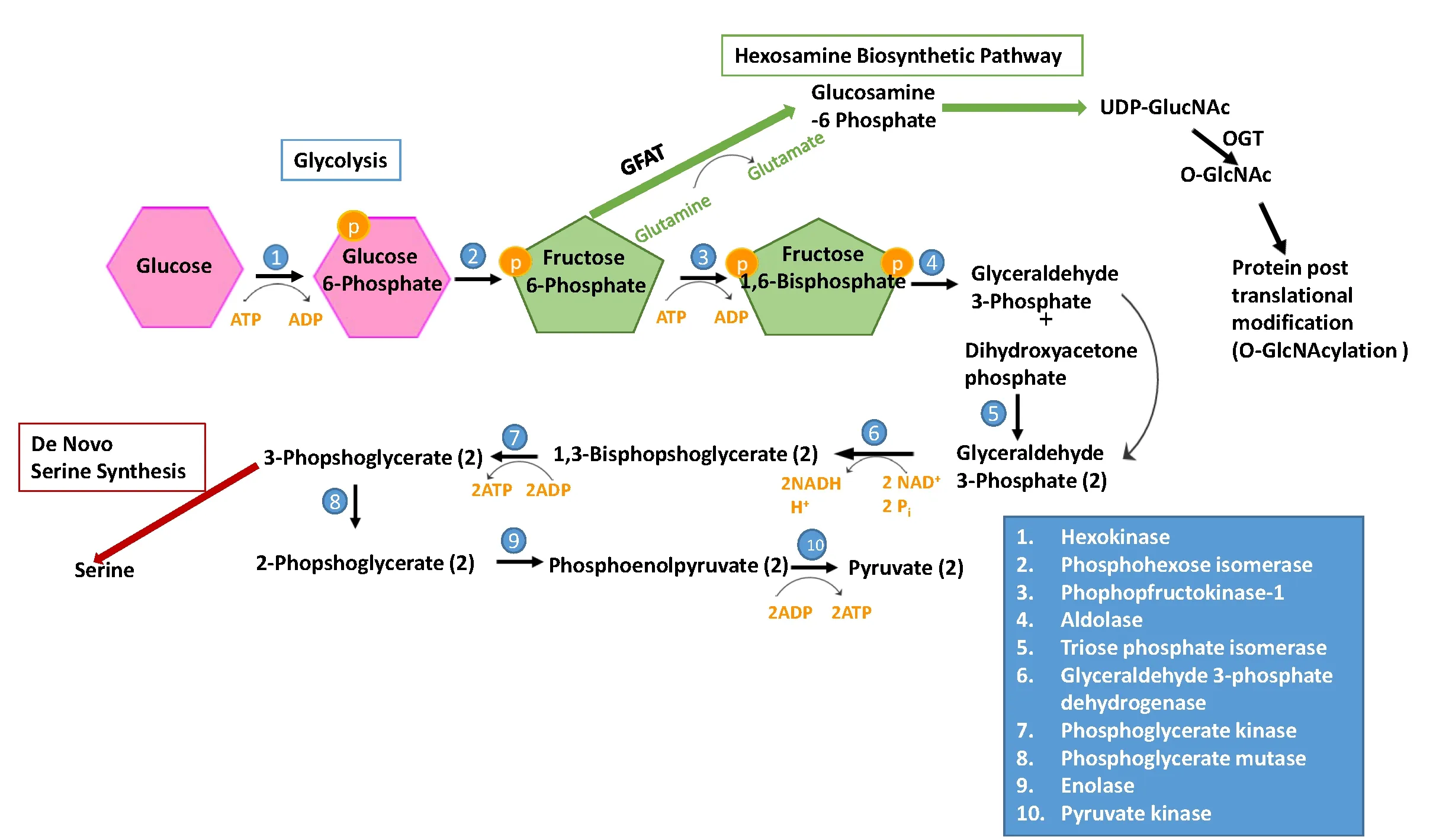
Figure 2 Glycolysis and other metabolic pathways in Müller Glia.

Figure 3 The unfolded protein response (UPR) and cellular metabolism.
Despite relying on a low-yield process for generation of ATP, Müller glia are sensitive to decreased access to ATP,and have been shown to contribute to acidification in the retina when ATP levels are low, via a sodium coupled bicarbonate transporter that can pump protons into the surrounding extracellular fluid (Tchernookova et al., 2018).This response has important implications for retinal function, because subtle changes in retinal pH can rapidly halt photoreceptor transmission. Retinal acidification, for example, has been shown to close photoreceptor calcium channels by reducing both chloride and calcium ionic currents (Barnes and Bui, 1991; Kleinschmidt, 1991). Additionally, although glycolysis is a cytosolic process, Müller glia are sensitive to problems with mitochondrial respiration. One of the most energy demanding functions of Müller glia is the removal of glutamate from the extracellular space, which is coupled to movement of Na+/H+/K+ions and powered by ATPase. Low ATP availability attenuates this function, increases glutamate in the extracellular space, leads to glutamate excitotoxicity, and eventually leads to death of inner retinal neurons.This is linked to pathology in diabetic retinopathy and glaucoma (Gurler et al., 2000).
The UPR
Protein folding is a critical and highly regulated cellular function for generation of mature and functional proteins with sophisticated secondary and tertiary structures, where the amino acid polymer is ‘folded’ and stabilized by hydrogen bonds, covalent bonds, and Van der Waals forces between residues (Christis et al., 2008). The organelle responsible for the majority of protein folding and packaging in the cell is the endoplasmic reticulum (ER). Additional protein folding takes place in cytosolic ribosomes and within the mitochondria (Gao et al., 2015). The ER works to regulate protein folding and maintain homeostasis through the monitoring of protein translation, folding, and degradation.Under homeostatic conditions, fully folded proteins are transported from the ER to the Golgi apparatus, where they can be secreted and transported to the appropriate location.However, when protein folding becomes disturbed, the ER employs the UPR to restore this critical function and eliminate the possibility of accumulation of harmful misfolded proteins in the ER, a condition known as ER stress. ER stress can be induced by a large variety of physiological factors and pathological conditions, such as environmental stress,increased demand for protein synthesis, hypoxia, and infl ammation, just to name a few. A frequent consequence of ER stress is misfolded protein aggregation. For most soluble proteins, folding establishes an energetically favorable conformation in which hydrophobic residues are oriented on the inside of the protein and hydrophilic residues are oriented on the outside (Christis et al., 2008; Gao et al., 2015; Hetz and Papa, 2018). The exposure of the hydrophobic residues in misfolded proteins leads to protein aggregation (Zhang et al., 2015; Christis et al., 2008).
The activation of the UPR is mediated by three transmembrane ER proteins: protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK), inositol requiring enzyme-1(IRE1), and activating transcription factor 6 (ATF6). These proteins contain an ER luminal domain that acts as a canonical sensor of ER stress; through this domain the proteins are bound to an ER chaperone namely glucose-related protein 78 (GRP78, also known as immunoglobulin binding protein,Bip) and are kept inactive in resting cells. Upon ER stress,GRP78 is sequestered by the misfolded or unfolded proteins and dissociates from the ER stress sensor proteins, resulting in the activation of the UPR (Figure 3).
The most conserved UPR branch is mediated by IRE1. Activated IRE1 splices a 26 nucleotide exon from the mRNA of a transcription factor named X-box binding protein 1 (XBP1).This process generates spliced XBP1 (XBP1s), which acts as a potent transcription factor to induce the expression of genes of ER chaperones and those encoding proteins involved in protein folding and ER-associated degradation (ERAD), a program that remove unfolded and misfolded proteins from the ER. In addition, XBP1 also regulates a diverse set of target genes that regulate metabolic processes including lipogenesis, gluconeogenesis and others (Gao et al., 2015; Piperi et al., 2016). Another important UPR branch that regulates the transcription of downstream UPR target genes is ATF6.ATF6 consists of a DNA binding domain as well as a luminal domain (Yamamoto et al., 2007). After dissociation from GRP78, ATF6 can travel to the Golgi apparatus via vesicular transport and undergo cleavage by two proteases, site 1 and site 2 proteases. The cleaved cytoplasmic portion of ATF6 is then activated, translocates to the nucleus, and regulates gene expression of ER chaperones, such as GRP78 and GRP94, and ERAD proteins (Sato et al., 2007; Zhang et al., 2015). A third and unique UPR branch is activated by PERK, a type-1 transmembrane kinase, through oligomerization and trans-autophosphorylation after dissociation from GRP78. This triggers phosphorylation of eukaryotic translation initiator factor-2(eIF2α), resulting in a reduction in the global protein translation and an increase in the production of ATF4, which in turn upregulates stress response genes, most prominently C/EBP homologous protein 10 (CHOP).
While the UPR is helpful to most organisms under normal conditions, it can be detrimental in disease conditions,where proteins are chronically misfolded due to mutations and/or environmental stressors. Several retinal diseases have been linked to increased ER stress and UPR activation(reviewed in Ma et al., 2014; Zhang et al., 2014; Gao et al.,2015; Chan et al., 2016). In retinitis pigmentosa for example,mutation of rhodopsin results in protein misfolding and a prolonged activation of the UPR that eventually leads to cell death (Athanasiou et al., 2017). Additionally, all three UPR branches have been implicated in regulation of pro-inflammatory factors, including vascular endothelial growth factor (VEGF), monocyte chemoattractant protein 1 (MCP-1), and α-crystallin-B (CRYAB) (Li et al., 2011; Zhong et al.,2012; Liu et al., 2013; Gao et al., 2015; Huang et al., 2015).These factors in turn promote pathological angiogenesis and increase vascular permeability, resulting in pathologies of diabetic retinopathy (Figure 3).
The UPR and Metabolic Pathways
Emerging evidence suggests a close interaction between the UPR signaling and metabolic pathways, both of which are believed to play a crucial role in cell adaptation and survival. On one hand, ER stress can be induced by changes in metabolism, including nutrient deprivation, hypoxia, and metabolic disorders (Hetz and Papa, 2018). In diabetes for example, insulin resistance leads to an increased need for insulin to respond to elevated blood glucose. This in turn increases the amount of insulin intermediates produced in pancreatic β cells, which challenges the ER folding capacity and leads to activation of the UPR (Hetz and Papa, 2018).On the other hand, the UPR has been shown to modulate metabolic pathways in a large variety of cell types and disturbance in the UPR components leads to metabolic diseases including obesity and diabetes (Feng et al., 2009; Sha et al.,2011; Yang et al., 2017). The regulation of lipid metabolism by the UPR and its implication in metabolic disease was recently reviewed by Ho and associates (Ho et al., 2018). In this section, we will focus on the role of the UPR in regulation of glucose metabolism and pathways related to retinal cells and Müller glia (Figure 3).
XBP1, which is induced during activation of the IRE1 branch of the UPR, is found to be a critical factor in regulation of glucose metabolism (Piperi et al., 2016). In liver cells and pancreatic cells, XBP1 regulates hepatic gluconeogenesis, glycogen production, and insulin secretion (Lee et al., 2011; Zhou et al., 2011). In cardiac tissue, activation of XBP1 protects against ischemic injury through regulation of the hexosamine biosynthetic pathway (HBSP) (Wang et al., 2014). The HBSP uses a glycolytic intermediate, fructose-6-phosphate (F6P) and glutamine to generate glucosamine-6-phosphate (Gln-6-P) and glutamate from the enzyme glutamine:fructose-6-phosphate aminotransferase(GFAT), which is found to be a direct target of XBP1. In addition, UDP-GlcNAc, the end product of the HBSP, can be converted by β-linked N-acetylglucosamine (O-GlcNAc)transferase (OGT) into O-GlcNAc for post-translational protein O-glycosylation (Marshall et al., 1991; Semba et al.,2014; Wang et al., 2014). In diabetic retinopathy, GFAT expression is upregulated by hyperglycemia, thereby increasing the O-GlcNAcylation of proteins (Gurel et al., 2013).Additionally, an increase in the heat shock protein (HSP)activity is linked to insulin resistance and diabetes (Marshall et al., 1991; Hawkins et al., 1997; Buse, 2006). A recent study reported that activation of the IRE pathway reduces glucose metabolism and mitochondrial respiration without inducing apoptosis in neuronal cells (van der Harg et al., 2017).It is hypothesized that IRE1 functions as a nutrition sensor and IRE activation during starvation induces the activation of XBP1, and its downstream target peroxisome prolifer-ator-activated receptor α (PPARα), which reprogram the mitochondria to switch from glucose to lipid metabolism thereby restoring energy homeostasis.
The ATF6 branch of the UPR has also been linked to cell metabolism but to a lesser extent. Glucose deprivation activates ATF6, which negatively regulates sterol regulatory element-binding protein 2 (SREBP2) and attenuates SREBP2-mediated lipogenesis (Zeng et al., 2004). Interestingly, overexpression of ATF6 increases abnormal lipid retention in the liver resulting in hepatic steatosis in mice through a physical interaction with PPARα (Chen et al.,2016b). In addition, activation of the ATF6-UPR arm is able to protect intestinal epithelial cells from mitochondrial damage induced by uncoupling oxidative phosphorylation with the H+ionophore, dinitrophenol (DNP) likely through induction of autophagy (Lopes et al., 2018). Deletion of ATF6α increases neuronal cell death caused by brain ischemia and this effect is believed, at least in part, mediated by reduced activation of astrocytes (Yoshikawa et al., 2015). In support of this finding, a recent study shows that ablation of ATF6α exacerbates ER stress-induced cell death of oligodendrocytes and increases demyelination in a mouse model of autoimmune encephalomyelitis (Stone et al., 2018). These finding support a role of ATF6 in maintaining the viability and function of glial cells in the central nervous system during pathologic and stress conditions. Whether this protective effect of ATF6 is associated with changes in cell metabolism and how ATF6 manipulation affect metabolic pathways warrant future investigation.
The PERK arm of the UPR has been shown to regulate cell metabolism through both mitochondrial and glycolytic pathways. A recent study reported that PERK enhances mitochondrial metabolism and protects mitochondrial morphology through promoting the stress-induced mitochondrial hyperfusion (SIMH) program (Lebeau et al., 2018).Deletion of PERK reduces glucose metabolism through downregulation of key glycolytic enzyme HK2 in glioma cells (Hou et al., 2015). In drosophila, ATF4 was found to be essential for upregulation of glycolytic enzymes and lactate dehydrogenase. These changes were thought to mediate a shift from oxidative phosphorylation based metabolism to one more heavily reliant on glycolysis or the Warburg effect(Lee et al., 2015). Furthermore, ATF4 can be activated by mitochondrial stress and is believed to function as a major regulator of mitochondrial stress response and could play an important role in mitochondria-related diseases (Quirós et al., 2017). Interestingly, several in vivo studies have shown that ATF4 expression is significantly increased in retinal neurons, Müller glia and vascular cells in animal models of diabetic retinopathy and autosomal dominant retinitis pigmentosa (Chen et al., 2012; Zhong et al., 2012; Bhootada et al., 2016). Overexpression of ATF4 in the retina results in enhanced inflammation, retinal dysfunction, and progressive neurodegeneration due to increased apoptosis and loss of photoreceptor cells (Huang et al., 2015; Bhootada et al.,2016). These data suggest that although ATF4 is essential for regulation of certain metabolic pathways and stress response, sustained induction or overexpression of ATF4 in disease conditions have detrimental effect on cell survival;however, whether ATF4 overexpression is suf ficient to cause metabolic disruption as seen in many neurodegenerative diseases remain elusive.
The UPR and Müller Glia Metabolism
Several studies have shown that the UPR is activated in retinal Müller cells under stressed conditions that cause metabolic disruption, such as low glucose, hypoxia, and oxidative stress(Devi et al., 2012; Wu et al., 2012; Zhong et al., 2012). Activation of the ATF4-CHOP pathway was found to be responsible for apoptosis and increased pro-in flammatory cytokine production from cultured Müller cells exposed to high glucose, hypoxia, or oxidized and glycated LDL (Wu et al., 2012;Zhong et al., 2012). In addition, Müller glia are considered highly susceptible to an increases in glucose, given that the cells primarily reply on glycolysis to generate ATP. The major glucose transporter in Müller glia is GLUT1, which is also the only glucose transporter carrying glucose across the BRB(Mantych et al., 1993; You et al., 2017). GLUT1 maintains the basal level of glucose in the cell in response to blood glucose, and its expression in the cell membrane corresponds to the cell’s metabolic activity (Kumagai et al., 1994). Thus, the function of GLUT1 is critical for maintaining energy metabolism in Müller glia. Interestingly, GLUT1 is also identified as a critical therapeutic target for metabolic disorders; inhibition of the transporter has been shown to attenuate diabetic retinopathy and diabetic complications (Lu et al., 2013; You et al., 2017). GLUT1 and GLUT2 are regulated by hypoxia inducible factor 1 (HIF1α), a transcription factor that has been shown to be directly regulated by XBP1 (Chen et al., 2001,2014). The regulation of GLUT1 and GLUT2 by HIF1α is well established across cell types and species, wherein increased HIF1α expression typically results in increases in GLUT1 and GLUT2 expression (Chen et al., 2001; Hayashi et al., 2004;Wan et al., 2017). Under hypoxia condition, HIF1α is upregulated to increase the production of cytokines that promote vascular permeability from Müller glia (Xin et al., 2013). The regulation of glucose transporters and glucose metabolism by the UPR in Müller glia is yet to be investigated.
In addition to glucose metabolism, several metabolic reactions occur in Müller glia using glycolytic intermediates, one of which is de novo serine synthesis. Serine is biosynthesized from 3-phsophoglycerate (3-PG), a 3 carbon sugar derived from glucose during step 7 of glycolysis. The 3-PG is converted to serine via 3 enzyme catalyzed steps. The enzymes,3-phosphoglycerate dehydrogenase (PHGDH), phosphoserine aminotransferase 1 (PSAT1), and phosphoserine phosphatase (PSPH), are all expressed in Müller glia (Zhang et al., 2018). This pathway is important for retinal homeostasis,because retinal neurons lack PHGDH and therefore rely on Müller glia for serine (Furuya, 2008). In cultured human Müller cells, disrupting serine synthesis through inhibition of PHGDH, or via oxidative stress does not significantly alter metabolism, but the combination of the two insults leads to metabolic disturbances, decreased mitochondrial ATP production, and cell death (Zhang et al., 2018). In addition,the disruption also results in an increase in the expression of ER chaperone HSP72. Proper function of HSP72 requires ATP hydrolysis. Given that ATP production is reduced, it is likely that the adaptive response of HSP72 is attenuated.This finding demonstrates a close interplay between protein homeostasis and metabolic stress.
Perspectives
There is emerging evidence to suggest an important role of the UPR in regulation of cellular metabolism, which in turn confers significant impact on cell viability, growth, and function. Relative to other tissues, there is little information available on how the UPR pathways are involved in regulation of retinal metabolism. Recently, we carried out a study to investigate how XBP1 deficiency in fluences retinal metabolism using a conditional knockout mouse line that lacks XBP1 in retinal neurons (McLaughlin et al., 2018). Our data suggests that loss of XBP1 has a profound effect on glycolysis in the retina. XBP1-deficient conditional knockout (cKO)mice demonstrate reduced retinal glycolysis, accompanied by significant structural degeneration and functional decline of the retina. In line with our results, a recent study shows that silencing XBP1 suppresses glycolysis and decreases ATP production in glioma cells (Liu et al., 2016). These findings support a regulatory role of XBP1 in glycolysis. However,how XBP1 regulates glycolysis in retinal neurons remains to be elucidated. Further, the potential implication of other UPR pathways, for example, IRE1, which is the upstream regulator of XBP1 activation, in regulation of metabolic pathways in retinal cells and maintenance of metabolic homeostasis of the retina is largely unexplored.
Among all types of retinal cells, Müller glia appear to an ideal cell to study the metabolic regulation because, as discussed above, these cells are the primary players in synthesizing and shuttling energy substrates, cytokines, trophic factors, and other regulatory proteins to surrounding cell populations. Specifically, this body of research would benefit from an exhaustive study of metabolic genes and proteins expressed in Müller glia. Understanding the amount of glycolytic substrates such as glucose-6 phosphate, fructose-6 phosphate, and 3-phosphoglycerate being shuttled to the pentose phosphate pathway, hexosamine biosynthetic pathway, and de novo serine synthesis pathway, respectively, is also important to inform where Müller glia ATP is derived from and allotted to, and how activity of the pathways will change in Müller glia in diseased conditions such as hypoxia and hyperglycemia. Last but not least, improvement in methods to be used for accurately measuring energy metabolism is critical for the success of this type of research. The recent development of Seahorse metabolic analyzers has enabled a real-time assessment of mitochondrial and glucose metabolism in cultured retinal cells and retinal tissues ex vivo. Future invention of new approaches to measure the actual metabolism in specific retinal cells including Müller glia in vivo would provide better tools for research to further understand the regulation of retinal metabolism in normal and diseases conditions.
Author contributions: KK, JJW, and SXZ wrote, revised and approved the final manuscript for publication.
Conflicts of interest: The authors declare that there are no conflicts of interest associated with this manuscript.
Financial support: This work is supported, in part, by NIH/NEI grants EY019949 and EY025061, and an Unrestricted Grant to the Department of Ophthalmology, SUNY-Buffalo, from Research to Prevent Blindness.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open access statement: This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike 4.0 License, which allows others to remix, tweak,and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewers: Giacinto Bagetta, University of Calabria, Italy;Rong-Kung Tsai, Buddhist Tzu Chi General Hospital, Taiwan, China; Steven Levy, Millennium Magnetic Technologies and MD Stem Cells, USA.
Additional file: Open peer review reports 1-3.
- 中国神经再生研究(英文版)的其它文章
- Restoration of an injured lower dorsal ascending reticular activating system in a patient with intraventricular hemorrhage
- Taurine protects against retinal and optic nerve damage induced by endothelin-1 in rats via antioxidant effects
- SIRT1 facilitates amyloid beta peptide degradation by upregulating lysosome number in primary astrocytes
- Cognitive deficits and Alzheimer-like neuropathological impairments during adolescence in a rat model of type 2 diabetes mellitus
- Enriched environment elevates expression of growth associated protein-43 in the substantia nigra of SAMP8 mice
- Achyranthes bidentata polypeptide protects dopaminergic neurons from apoptosis induced by rotenone and 6-hydroxydopamine