
基于染色质免疫共沉淀的高通量测序数据集的顺式调控模体发现算法
2018-08-28 08:53:08冯艳霞张志红张少强
计算机应用 2018年6期
冯艳霞,张志红,张少强
(天津师范大学计算机与信息工程学院,天津300387)
(*通信作者电子邮箱zhangshaoqiang@tjnu.edu.cn)
0 引言
基于新一代基因测序的高通量技术,特别是染色质免疫共沉淀的高通量测序(ChromatinImmunoprecipitation Sequencing,ChIP-Seq)技术[1],在全基因组范围内的使用,很大程度上改变了生物学家定位大规模真核基因组顺式调控元件和模体的方式[2]。顺式调控模体是同一转录因子结合位点的序列集合。模体发现问题一直是生物信息学比较重要的研究课题之一。模体所蕴含的遗传信号,对于在全基因组范围内研究基因的转录与调控机制具有重大意义。在ChIP-Seq技术之前的模体发现,主要是通过比较同源基因或共调控基因非编码区域的保守性来预测调控元件模体,其主要方法包括:通过多序列比对寻找保守序列片段;通过位置赋权矩阵更新来遍历调控元件;运用统计取样的估计方法等。这些方法均取得了较好的预测结果,并应运而生了大量模体发现算法和工具,例如多重期望最大模体提取(Multiple Expectation maximization for Motif Elicitation,MEME)算法[3]、MotifSampler算法[4]、Weeder算法[5]等,为生物学家对真核生物全基因组[6]的解读和研究提供了有利保障,推动着生物信息学的蓬勃发展。模体发现算法的工作流程如图1所示。
由于ChIP-Seq单次实验产生了海量的碱基读序(reads),通过序列比对拼接及结合峰预测[7]后仍有成千上万条序列,这比传统的同源基因或共调控基因序列规模大很多。为了处理如此大规模的数据,近来的算法主要采用“有区别的”(discriminative)模型[8]策略,主要包括:一是选择数据集的子集作为实验的输入;二是在消耗较大数据存储空间的情况下,运用新型数据结构来节省运行时间;三是将实验数据集分割成两个不等的部分,即实验组和对照组,仅将实验组作为模体发现算法的核心运行数据,然后根据实验组发现的模体,与对照组进行比对。上述第一种方法数据损失较多,要预测的结合位点模体信息在子集中可能不保守,从而大大影响算法准确性,例如独立成分分析(Independent Component Analysis,ICA)和套层抽样相结合的的模体发现算法——NestedMICA(Nested saMpling and ICA)[9];第二种方法的主要问题是随着数据规模增加,会成倍增加内存需求和时间复杂性,从而降低了算法运行速度,例如有区别的正则表达式模体提取(Discriminative Regular Expression Motif Elicitation,DREME)算法[10];相比较而言,第三种方法更注重序列数据集的分隔方法,但不恰当的数据分隔可能导致错误的结果,例如Amadeus[11]、Trawler[12]。特别是,随着 ChIP-Seq 数据的海量增长,其噪声也越来越多,从而导致预测结果具有较高的错误发生率(False Discovery Rate,FDR)。此外,模体发现算法在追求时间与空间的高效外,还应准确识别出模体的真实长度。
为了解决上述问题,本文设计了一个基于ChIP-Seq数据集的顺式调控模体发现算法——FisherNet,该算法既能提高模体预测的准确率,又能提高算法运行速度。
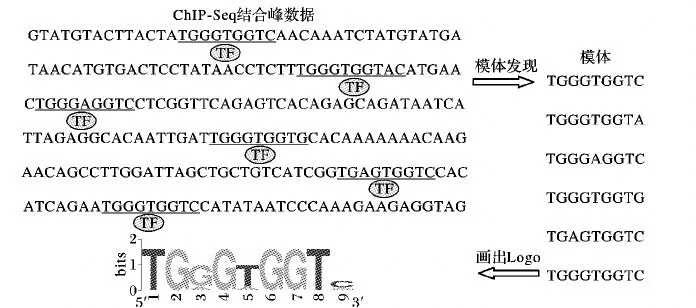
图1 模体发现流程Fig.1 Flow chart of motif finding
1 数据来源与预处理
为了检验算法的优越性,FisherNet算法使用与DREME[10]算法相同的数据集,包括13个小鼠胚胎干细胞(Mouse Embryonic Stem Cell,mESC)ChIP-Seq 数据集[13]、3 个小鼠红细胞数据集和1个人类淋巴母细胞系数据集。mESC数据集为12个转录因子,它是维持多能性和CTCF的关键。小鼠红细胞的数据集为 Gata1[14]、Klf1[15]和 Tal1,是红细胞生成的关键调节者。人体淋巴细胞数据集是维生素D受体包括在使用骨化三醇的刺激前和之后的ChIP-Seq数据。另外,基于人的 ENCODE(Encyclopedia of DNA Elements)[16]数据库的ChIP-Seq数据集规模超大,本文选取其中的10个有已知模体的转录因子(ATF1、ATF3、CTCF、E2F4、EBF1、ECF1、POL2、ZNF274、USF2、YY1)数据集进行比较。这些数据集源文件格式为FASTQ格式。
2 算法的主要步骤
本文算法的流程如图2所示。
2.1 生成背景数据集
FisherNet算法采用了三阶马尔可夫链[17]生成背景数据,即遍历实验组ChIP-Seq数据集统计每三个碱基短序列(共43=64种)后出现各个碱基的频率得到概率转移矩阵。然后用该转移矩阵生成与实验组数据中序列长度和数目相同的背景序列数据集。
2.2 遍历短序生成哈希表
为了统计每个长度为k的短序(k-mer)[18]在各个序列是否存在,FisherNet分别遍历实验数据集和背景数据集中的每条序列并用两个哈希表分别存储所有的k-mer信息。哈希表的键为k-mer,键值为该k-mer所在序列编号集合。因为真核生物的结合位点长度主要介于4~8个碱基长。FisherNet算法默认k从4到8个碱基为窗口长度分别进行遍历。在遍历过程中,同时考虑k-mer在序列的互补链是否存在,若存在,则将该序列的编号也放到该k-mer键值对应的集合中。遍历过程如图3所示。

图2 本文算法流程Fig.2 Flow chart of proposed algorithm
2.3 根据费舍尔精确检验计算P值
FisherNet算法采用费舍尔精确检验分析每个k-mer的富集情况[19]。对于每个k-mer,通过哈希表查询记录下它在实验数据集和背景数据集中出现的序列条数。如表1所示,a代表某k-mer在实验组数据集中出现过的序列数,b代表该短序在背景数据集中出现过的序列数,相对地,c和d分别代表该短序在两组实验数据集中未出现的序列数。
根据表1通过超几何分布公式计算P值[20]为:

若P值大于意义临界值0.05(假设检验常规设P值的阈值为0.05,在算法中该阈值作为输入参数可由使用者自主调整),说明该条DNA短序列存在基因信息与背景差异较小,则对其不再考虑。上述计算P值的式(1),对于小规模数据集效果良好,对于大型ChIP-Seq数据集,则阶乘n!会是超大的数,甚至超出计算机整数计数范围。对此,FisherNet引入了斯特林(Stirling)公式,对组合数的计算先取其对数ln,再取指数。斯特林如式(2)所示,与ln C推导如式(3)、(4)所示:
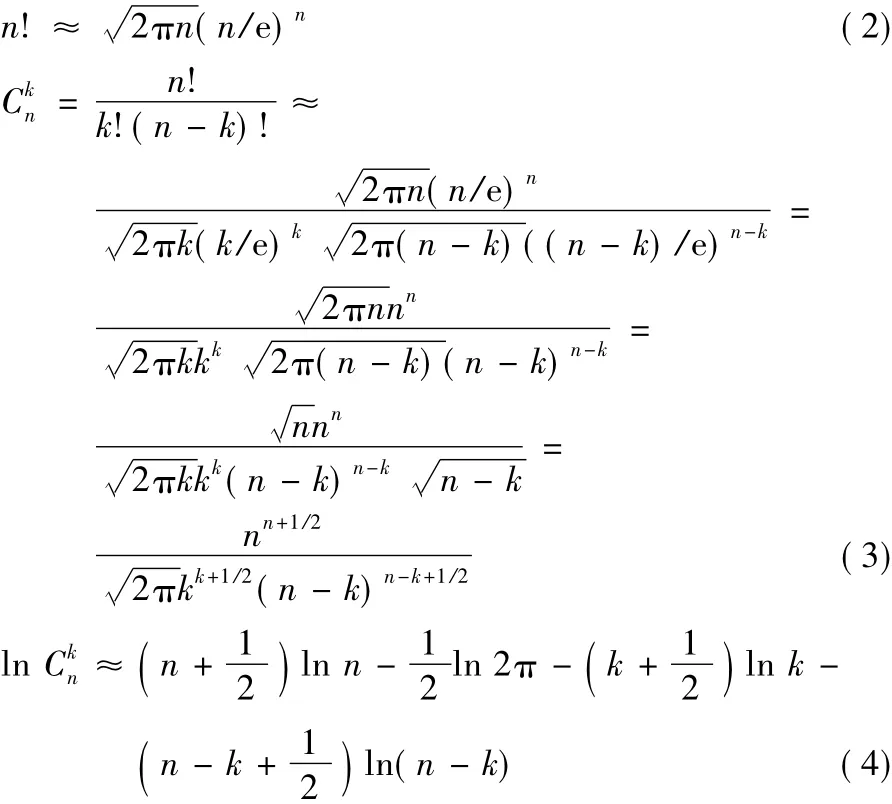
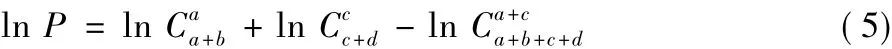
则P值公式就等价于:
对式(1)两边取对数得:
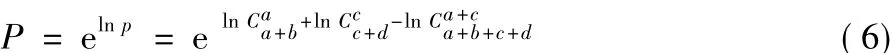

对于每个k-mer,用式(6)计算其P值,若P值小于0.05,以此k-mer为键,其P值为键值保存到一个新的哈希表。通过比较各k-mer的键值,对k-mer进行排序得到候选k-mer集合。

表1 k-mer(模体)序列在实验组和背景组的数据统计Tab.1 Statistics of k-mer(motif)sequence in experimental group and background group
2.4 构建初始模体
在候选k-mer集合中选取P值最小的k-mer作为种子,依次向下遍历与种子长度相同且只有一个位置不同的k-mer,将其合并形成初始模体。每合并一个k-mer就要通过计算出现模体和未出现模体的序列数来更新费舍尔精确检验表并计算合并后模体的P值,若P值 >0.05,则停止k-mer合并。统计初始序列上各碱基的出现频率得到位置频率矩阵P=(P(b,i))4×k[21],其中 P(b,i)表示碱基 b 在位置 i出现的频率;然后再根据式(7)计算出对应的位置赋权矩阵W =(W(b,i))4×k[22],其中:P(b)是碱基 b 在实验数据集上出现的频率(为了能够计算对数值,当P(b,i)=0,令P(b,i)=10-5)。构建初始模体的流程如图4中第① ~③步所示。
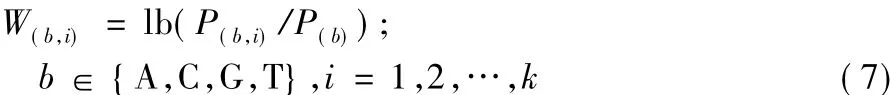
2.5 生成最终模体
用上述生成的位置赋权矩阵扫描候选k-mer集合中的每一个相同长度的k-mer={b1,b2,…,bk},为了衡量一个k-mer是否属于模体,本文用式(8)(公式中的Smax、Smin分别为位置赋权矩阵中每列元素的最大值的和与最小值的和)计算k-mer的相对分数值R并只保留R≥0.80(根据已知模体统计有80% 的位置具有保守性)的k-mer,以此生成最终模体。如图4中第④到⑤步所示。
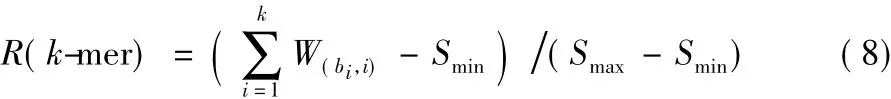
3 实验与结果分析
FisherNet算法运用mESC的ChIP-Seq数据集和一个意义临界值对结果进行具体分析。如表2为FisherNet算法预测出的模体发现情况。将算法发现的模体序列与Tomtom数据库[23]中的真实模体进行比较,针对Tomtom标准数据库中已有的13个重要模体,FisherNet算法发现了其中的10个。
此外,针对mESC ChIP-Seq数据集,本文分别运行了其他六种被广泛应用且能发现辅调控因子模体的模体发现算法,即 MEME[3]、Weeder[5]、NestedMICA[9]、DREME[10]、Amadeus[11]、Trawler[12],并将这些算法的预测结果与 FisherNet顺式调控模体算法相比较,统计结果如表3所示。其中:N为各算法发现的模体总数;S为与Tomtom标准数据库中相匹配的模体数;C为算法发现的辅调控因子模体数。在发现转录因子模体上FisherNet与 DREME、Weeder、NestedMICA 和 MEME 均达到最高,但在发现辅调控因子模体上FisherNet算法要优于其他算法。这说明该算法在保证发现模体的精确度的同时,能发现更多的辅调控因子模体,FisherNet总体能够达到80%(即(6.1+10)/20)的精确度。表3最后一列为各算法对13个mESC数据集的平均运行时间,可以看出,FisherNet是所有比对算法中平均运行时间最短的,而NestedMICA和Weeder是运行时间最长的两个算法。

表2 小鼠的ES细胞ChIP-Seq数据集发现的模体Tab.2 Found motif in mouse ES cells ChIP-Seq dataset

表3 不同模体发现算法结果统计Tab.3 Results statistics of different motif finding algorithms
由于DREME是MEME开发者设计的改进算法,为了进一步检验除了MEME、Weeder和NestedMICA之外的四种算法的效率,分别取序列规模为1兆、2兆、3兆和4兆碱基对的数据集进行运算,图5为四种算法的运行时间评估折线图。由图5可以看出,与其他算法相比,随着数据集序列碱基规模的增加,FisherNet算法运行时间最短且增加幅度不大,充分说明了该算法在运行效率上的优越性。将上述算法应用到选取的ENCODE[16]数据集中,发现每一个真实模体均存在于FisherNet算法输出的前10个模体中,而其余算法的前10个模体中含有真实模体的准确度均小于80%。

图5 不同算法运行时间比较Fig.5 Running time comparison of different motif finding algorithms
4 结语
本文基于ChIP-Seq数据的模体发现,以ChIP-Seq结合峰数据集为算法输入数据,提出一个全新的顺式调控模体发现算法(本文的FisherNet由 Perl语言编写,代码下载地址为http://www.escience.cn/people/sqzhang/program.html.)。该算法运用费舍尔假设检验来寻找区分度高的k-mer并通过位置赋权矩阵进行优化筛选。与其他多种模体发现算法进行比较,验证了该算法大大提高了模体发现的数目和准确度,成功预测到许多核心模体及辅调控因子模体。该算法目前只对几种代表性的ChIP-Seq数据进行了验证,对其他物种的数据是否具有鲁棒性有待进一步验证。
猜你喜欢
中国西部(2022年2期)2022-05-23 13:28:20
计算机与现代化(2021年5期)2021-05-27 06:51:34
南大法学(2021年6期)2021-04-19 12:27:30
教学考试(高考生物)(2020年6期)2020-11-23 05:25:56
食品与生物技术学报(2020年8期)2020-01-06 08:00:56
活力(2019年15期)2019-09-25 07:22:12
科学24小时(2019年5期)2019-06-11 08:39:38
发明与创新(2019年9期)2019-03-26 02:22:48
智能计算机与应用(2019年1期)2019-01-11 06:03:06
测控技术(2018年6期)2018-11-25 09:50:24
