
腺嘌呤葡萄糖苷的合成
2018-08-23 12:10:34孙莉萍孙明明渠桂荣
精细石油化工 2018年4期
孙莉萍,孙明明,夏 然*,渠桂荣
(1. 新乡学院 生命科学技术学院,河南 新乡 453003;2. 新乡学院化学化工学院,河南 新乡 453003; 3. 河南师范大学化学化工学院,河南 新乡 453007)
腺嘌呤葡萄糖苷[1,9-(β-D-glucopyranosyl)adenine,CAS:3181-39-3]是天然核苷腺苷的类似物,是将腺苷的核糖改变为葡萄糖[1],是最早被研究的嘌呤类吡喃糖苷类化合物。研究发现,腺嘌呤葡萄糖苷具有抑制多种癌细胞增生[2]、抑制转运蛋白NupC和NupG等活性[3]。还可以嵌入RNA链中,用于碱基识别和寡苷酸生理活性的研究[4]。多种具有良好活性的核苷类抗生素也具有腺嘌呤葡萄糖苷的结构片段。同时,以腺嘌呤葡萄糖苷为底物进行结构修饰,得到多样性的嘌呤类吡喃糖苷类化合物,也得到了广泛关注。
目前腺嘌呤葡萄糖苷的合成方法主要有:1)1-溴乙酰基葡萄糖和苄氨基嘌呤的汞盐缩合,脱除乙酰基和苄基得到[5];2)在回流和四氯化锡存在下,乙酰基保护的葡萄糖和硅醚保护的腺嘌呤缩合[6],脱除保护基得到;3)苄基保护的胞嘧啶葡萄糖苷和腺嘌呤在TMSCl(三甲基氯硅烷)存在下,在吡啶中回流发生碱基转移,然后脱除保护基得到[7]。这些方法存在以下不足:1)用到过量四氯化锡、氯化汞等重金属盐,毒性大,成本高;2)温度较高,易产生无活性的α异构体,分离提纯困难;3)原料合成步骤多,不易得。由于1的合成难度大,限制了其应用范围。
本研究以易于制备的1-氯-2,3,4,6-四-O-乙酰基葡萄糖(5)代替1-溴-2,3,4,6-四-O-乙酰基葡萄糖,在温和条件下和硅醚化的6-氯嘌呤(3)缩合,脱除乙酰基后得到腺嘌呤葡萄糖苷,共4步,总收率80%,并用1H NMR、13C NMR和HRMS确证腺嘌呤葡萄糖苷和6的结构。该法条件温和,原料易得,为腺嘌呤葡萄糖苷提供了较好的合成方法。合成路线见图1。

图1 合成路线
1 实 验
1.1 原料与仪器
6-氯嘌呤、β-D-(+)-五乙酰基葡萄糖,分析纯,洛阳德胜生物科技有限公司;其他所用试剂均为市售分析纯;乙腈提前用氢氧化钾干燥处理。
AC 400型核磁共振仪(DMSO-d6或CDCl3为溶剂,TMS为内标),德国Bruker公司;Q-TofMS/MS型高分辨质谱仪,美国Waters公司;XRC-1型显微熔点仪(温度计未校正),四川大学科仪厂。
1.2 合成方法
1.2.16-氯-9-(三甲基硅基)-9H-嘌呤(3)的合成
6-氯嘌呤(0.77 g,5 mmol)和HMDS(40 mL,六甲基二硅胺烷,分析纯)加入到三口瓶中,加入硫酸铵(0.1 g,0.75 mmol),加热回流反应10 h,60 ℃减压(真空度:0.09 MPa)浓缩,得到淡黄色油状物3(1.1 g,98%)。
1.2.21-氯-2,3,4,6-四-O-乙酰基葡萄糖(5)的合成
β-D-(+)-五乙酰基葡萄糖(2.0 g,5 mmol),加入到100 mL三口瓶中,加入二氯甲烷(50 mL),搅拌10 min使之溶解,冷却至0 ℃,滴加乙酰氯(0.5 mL),通入干燥的氯化氢气体(0.9 g,25 mmol),室温搅拌反应5 h。反应液真空浓缩(温度不高于40 ℃),用甲苯(5 mL)共沸三次,以除去残留溶剂,得到无色油状物5(1.8 g,99%)。
1.2.31-(6-氯-9H-嘌呤基)-2,3,4,6-四-O-乙酰基葡萄糖(6)的合成
上述3和5转移到三口瓶中,加入乙腈(30 mL)和四氯化锡(2%),60℃搅拌30 min,TLC[展开剂:V(甲醇)∶V(二氯甲烷)= 1∶9),Rf=0.29],加入H2O(5 mL),继续搅拌10 min,过滤,滤饼用乙腈(5 mL)淋洗,滤液减压浓缩(真空度0.09 MPa),浓缩物中加入二氯甲烷(20 mL)和水(20 mL),充分搅拌20 min,分出有机相,水相用二氯甲烷(20 mL)萃取,合并有机相,分别用饱和碳酸氢钠溶液(20 mL)和饱和氯化钠溶液(20 mL)洗涤,无水硫酸钠干燥,减压浓缩,得到淡黄色油状物6(2.1 g,87%)。纯度>98.0% [HPLC归一化法:色谱柱Kromasil C18柱(4.6 mm×150 mm,5 μm);流动相V(水)∶V(乙腈)=1∶1);检测波长254 nm;流速 1 mL/min;进样量 10 μL]。
1H NMR(CDCl3, 400 MHz),δ:8.85 (s, 1H), 8.77 (s, 1H), 6.60 (d,J=5.2 Hz, 1H), 6.10 (t,J=8.4 Hz, 1H), 5.44~5.40 (m, 1H), 5.30 (t,J=9.6 Hz, 1H), 5.22(t,J=8.4 Hz, 1H), 4.32~4.23 (m, 1H), 4.16~4.02 (m, 1H)。13C NMR(CDCl3, 100 MHz),δ:170.5, 170.0, 169.3, 169.1, 168.9, 152.4, 143.1, 131.4, 118.8, 91.6, 77.2, 72.7, 67.7, 61.4, 20.7, 20.6, 20.5。HRMS(C19H22ClN4O9)485.107 0 [M+H]+。
1.2.4腺嘌呤葡萄糖苷(1)的合成
NH3/MeOH氨解法:上述6加入到高压反应罐中,加入饱和NH3/MeOH溶液(100 mL),密封,100 ℃反应10 h(反应罐压力0.2 MPa),冷却至室温,缓慢释放压力,反应液减压浓缩,得到淡黄色油状物,加入无水乙醇(20 mL),加热溶解,加入活性炭(0.1 g)脱色,趁热过滤,滤液减压浓缩,得到白色固体1(0.95 g,收率79%)。
一锅法:上述6加入到高压反应罐中,用MeOH(50 mL)溶解,加入Na2CO3(5.3 mg),室温搅拌1 h,TLC检测原料6消失,再加入饱和NH3/MeOH溶液(50 mL),密封,100 ℃反应10 h(反应罐压力0.1 MPa),冷却至室温,缓慢释放压力,反应液减压浓缩,得到淡黄色固体,加入无水乙醇(20 mL),加热溶解,加入活性炭(0.1 g)脱色,趁热过滤,滤液减压浓缩,得到1(1.2 g,收率92%)。纯度>98%[HPLC归一化法:流动相V(水)∶V(乙腈)=3∶7,其他条件与化合物6一致]。
白色固体,m.p. 222~224 ℃(文献[8]:223~225 ℃)。1H NMR(400 MHz, DMSO-d6),δ8.31(brs, 1H), 8.14 (s, 1H), 7.28 (brs, 2H), 5.40 (d,J= 9.6 Hz, 1H), 5.20 (brs, 1H), 4.63 (t,J=0.4 Hz, 1H), 3.99 (d,J=11.2 Hz, 1H), 3.45~3.35 (m, 5H), 3.24 (d,J= 8.8 Hz, 1H)。13C NMR (100 MHz, DMSO-d6),δ:156.4, 153.1, 150.3, 140.2, 119.1, 83.2, 80.4, 72.7, 72.6, 70.2, 63.4。HRMS(C11H15N5NaO5): 320.096 5 [M+Na]+。
2 结果与讨论
由于该合成路线关键步骤在于糖苷键的生成,本实验以6-氯嘌呤和β-D-(+)-五乙酰基葡萄糖的缩合为模板反应,考察催化剂、溶剂、投料比、反应时间、反应温度及反应规模对缩合产物收率的影响。
2.1 催化剂对收率的影响
以6-氯嘌呤和β-D-(+)-五乙酰基葡萄糖的缩合反应为模板反应,以核苷碱基和糖缩合反应时常用的1,2-二氯乙烷为溶剂,改变催化剂的种类和投加量,考察催化剂及用量(摩尔分数)对缩合产物收率的影响。结果见表1。
由表1可知,催化剂对缩合产物收率有较大的影响。当使用三甲基氯硅烷和氯化汞时,收率分别为37%和29%。催化剂改变为四氯化锡时,收率提高到70%。但是因为四氯化锡的使用导致后处理产生絮状沉淀,所以尝试降低四氯化锡的投加量。当四氯化锡投加量(摩尔分数,下同)降低为2%时,收率可以达到78%。另外,不加催化剂时,收率也可以达到62%。所以综合考虑较佳的四氯化锡用量为2%。
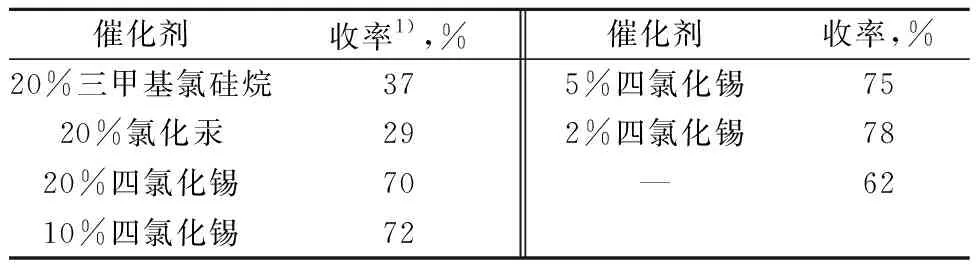
表1 催化剂对缩合产物收率的影响
注:6-氯嘌呤(0.77 g,5 mmol),β-D-(+)-五乙酰基葡萄糖(2.0 g,5 mmol),1,2-二氯乙烷(20 mL),室温,1 h;
1) 分离收率;下同。
2.2 溶剂对收率的影响
以6-氯嘌呤和β-D-(+)-五乙酰基葡萄糖的缩合为模板反应,以2%的四氯化锡为催化剂,改变溶剂的种类,考察溶剂对缩合产物收率的影响。结果见表2。
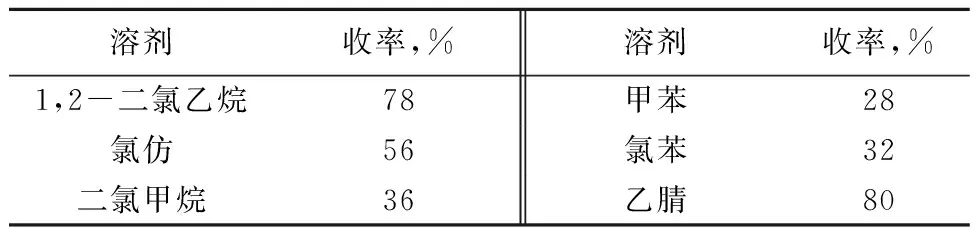
表2 溶剂对收率的影响
注:6-氯嘌呤(0.77 g,5 mmol),β-D-(+)-五乙酰基葡萄糖(2.0 g,5 mmol),溶剂(20 mL),四氯化锡(2%),室温下反应1 h。
由表2可知,当溶剂为1,2-二氯乙烷时,收率为78%。氯仿和二氯甲烷收率分别为56%和36%。当溶剂改变为极性更小的甲苯和氯苯时,收率降低到28%和32%。由此可见溶剂的极性降低对反应不利。当溶剂为乙腈时,收率可以达到80%。所以选择乙腈为较佳溶剂。
2.3 投料比对收率的影响
以6-氯嘌呤为原料,改变β-D-(+)-五乙酰基葡萄糖的投加量,以2%的四氯化锡为催化剂,以乙腈为反应溶剂,考察投料比对缩合产物收率的影响,结果见表3。
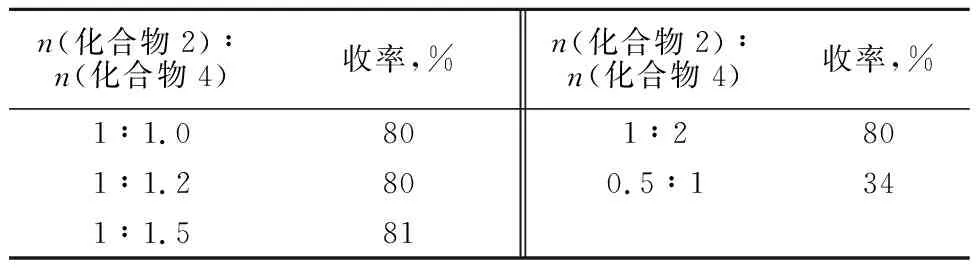
表3 反应规模对收率的影响
注:6-氯嘌呤(0.77 g,5 mmol),乙腈(20 mL),四氯化锡(2%),室温下反应1 h。
由表3可知,当n(化合物2)∶n(化合物4)=1∶1时,收率为80%。当n(化合物2)∶n(化合物4) =1∶1.5和1∶2时,收率基本不变,但是反应时间有所缩短。综合考虑收率及成本因素,以n(化合物2)∶n(化合物4)=1∶1为较佳。
2.4 反应时间对收率影响
以6-氯嘌呤和β-D-(+)-五乙酰基葡萄糖的缩合反应为模板反应,以2%的四氯化锡为催化剂,以乙腈为反应溶剂,考察反应时间对缩合产物收率的影响,结果见表4。由表4可知,较佳反应时间为2 h。

表4 反应时间对收率的影响
注:6-氯嘌呤(0.77 g,5 mmol),β-D-(+)-五乙酰基葡萄糖(2.0 g,5 mmol),乙腈(20 mL),四氯化锡(2%),室温。
2.5 反应温度对收率的影响
以1∶1 的6-氯嘌呤和β-D-(+)-五乙酰基葡萄糖的缩合为模板反应,以摩尔分数为2%的四氯化锡为催化剂,以乙腈为反应溶剂,反应2 h,考察反应温度对缩合产物收率的影响,结果见表5。
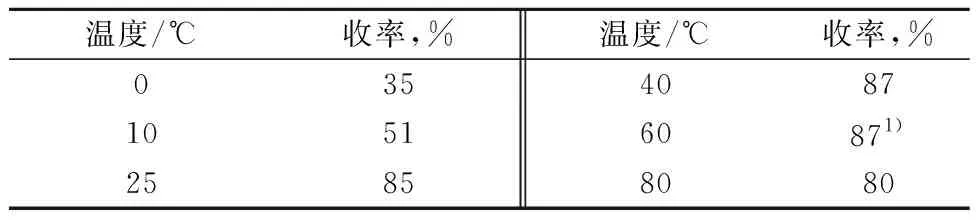
表5 反应温度对收率的影响
注:6-氯嘌呤(0.77 g,5 mmol),β-D-(+)-五乙酰基葡萄糖(2.0 g,5 mmol),乙腈(20 mL),四氯化锡(2%),2 h;
1) 反应时间为0.5 h。
由表5可知,在低温条件下,反应速度慢,收率低。当反应温度为0 ℃和10 ℃时,收率仅35%和51%。升高反应温度,产物的收率增加。反应温度为室温(25 ℃)时,收率达到85%。继续升高反应温度,收率不变,但是反应速度加快,反应温度为60 ℃时,反应时间仅需30 min。综合考虑,较佳反应条件为60 ℃。
2.6 反应规模对收率的影响
以1∶1的6-氯嘌呤和β-D-(+)-五乙酰基葡萄糖的缩合反应为模板反应,以2%的四氯化锡为催化剂,60 ℃反应0.5 h,改变反应规模,考察反应规模对缩合产物收率的影响,结果见表6。

表6 反应规模对收率的影响
注:n(6-氯嘌呤)∶n(β-D-(+)-五乙酰基葡萄糖)=1∶1,乙腈,四氯化锡(2%),60℃,0.5 h。
由表6可知,随着反应规模的扩大,收率稍有降低。反应规模为10 g时,收率82%。当反应规模达到200 g时,收率仍然可以达到75%。至于为什么随着反应规模的扩大收率稍有降低,还需要在下一步的深入研究中考察。
2.7 氨解条件的优化
缩合物6在NH3/MeOH溶液中高压反应,在脱除糖环上的乙酰基的同时,使6-Cl原子氨解。但是同时产生4倍量的乙酰胺,乙酰胺溶解在溶剂中,使产物难以析出,收率仅79%。为了解决这个问题,先加入5 mol%的Na2CO3,室温搅拌1 h,脱除乙酰基,然后不需要分离,直接加入饱和NH3/MeOH溶液,高压氨解,得到产物,这样避免了产生过量的乙酰胺,产物1很容易从反应体系中析出,不需要柱层析,为进一步的应用创造了条件。

图2 氨解条件的选择
3 结 论
以6-氯嘌呤和β-D-(+)-五乙酰基葡萄糖为原料,以4步和80%的总收率得到腺嘌呤葡萄糖苷。考察了催化剂、溶剂、投料比、反应时间、反应温度及反应规模对关键中间体收率的影响。缩合反应的反应规模可以扩大到200 g,收率达75%。改善了脱除乙酰基和氨解的方法,使产物易于结晶,不需要柱层析。该方法避免了传统方法中需要大量重金属盐催化剂及高温等苛刻条件,原料廉价易得,操作简便,为腺嘌呤葡萄糖苷及其类似物的合成提供了有益参考,具有一定的应用前景。
猜你喜欢
陕西师范大学学报(自然科学版)(2022年5期)2022-11-09 10:42:10
云南化工(2021年8期)2021-12-21 06:37:20
云南化工(2020年4期)2020-05-19 09:15:24
科海故事博览·中旬刊(2020年3期)2020-03-15 05:49:04
基层中医药(2018年8期)2018-11-10 05:32:00
安徽化工(2018年5期)2018-10-23 03:22:34
益寿宝典(2018年17期)2018-01-26 15:44:57
食品工业科技(2014年19期)2014-07-25 06:17:42
天然产物研究与开发(2014年6期)2014-04-27 14:16:02
中国海洋大学学报(自然科学版)(2014年12期)2014-02-28 12:22:04
