
月见草油气相色谱指纹图谱研究
2018-08-10 02:17:04王慧竹徐丽华
中国酿造 2018年7期
王慧竹,陈 帅*,石 琳,韩 绪,张 龙,张 敏,徐丽华
(1.吉林化工学院 化学与制药工程学院,吉林 吉林 132022;2.吉林大学 药学院,吉林 长春 130021)
月见草(Oenothera biennisL.)为柳叶菜科月见草属1~2年或多年生草本植物,其原产于北美地区,在我国主产于东北三省[1],月见草全身是宝,月见草挥发油可食用,花可提取芳香油,茎皮纤维可制绳,根是解热、消炎的良药,茎和叶有清热解毒、镇痛的功效[2-3],种子可榨油食用和药用,其中月见草油是其主要成分,主要由亚油酸、油酸、棕榈酸、亚麻酸多种脂肪酸和维生素等组成[4],具有良好的降脂、降糖、减肥、抗炎、抗氧化、抗血栓功能[5-7],对老年慢性疾病具有医疗保健作用。随着对月见草油药理作用的进一步认识,全世界对月见草油的需求也日益增长。目前对月见草油的研究主要集中在提取工艺、成分分析、药理作用等方面[8-10]。
中药指纹谱图技术是指用色谱、光谱检测方法获得中药成分的谱图,然后对图谱进行“过滤”、简化等加工处理,从而获得稳定和专属的特征指纹图谱,以此来控制药材质量[11-13]。
本实验以月见草油为研究对象,采用气相色谱法结合柱前甲酯衍生化法,建立不同产地月见草油的指纹图谱,通过国家药典委员会指纹图谱评价软件计算相似度,并结合化学计量学中的聚类分析和主成分分析,探讨了不同批次月见草油差异的来源,以期为月见草油的质量控制提供科学依据。
1 材料与方法
1.1 材料与试剂
来源于不同产地的月见草药材购自吉林各大药材零售商,经吉林化工学院陈帅副教授鉴定为月见草正品,月见草药材的批次编号、批号及产地信息汇总情况见表1。
石油醚、环己烷(分析纯)、对照品:γ-亚麻酸甲酯(批号:20110723,纯度≥98%)、亚油酸甲酯(批号:20176307,纯度≥98%)均由武汉华士特工业生物技术开发有限公司提供。
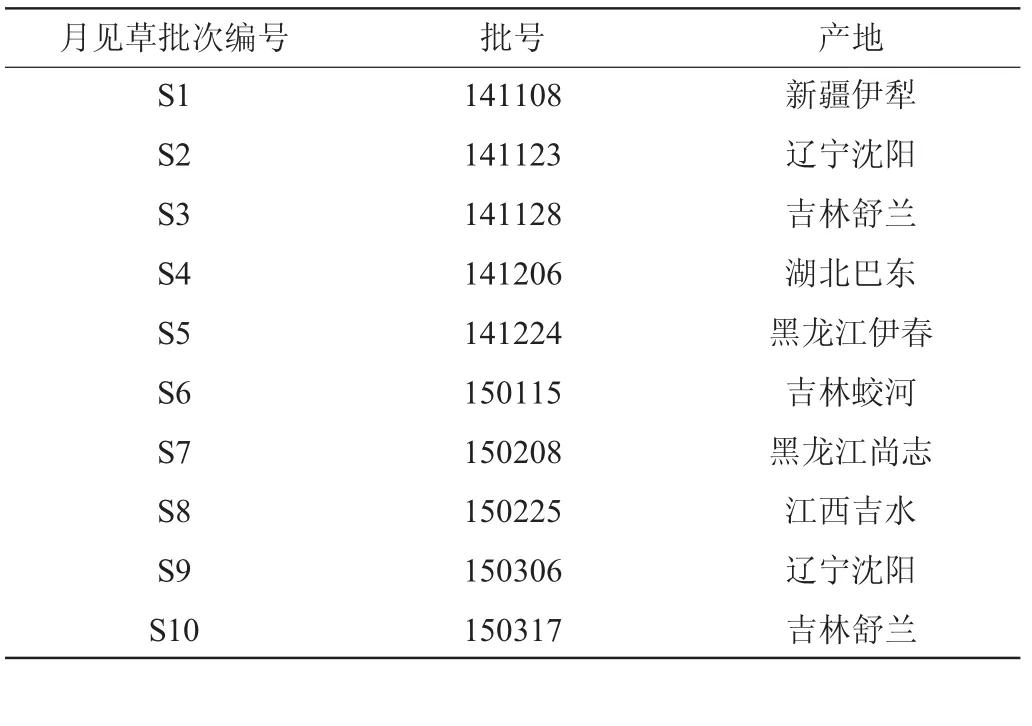
表1 月见草来源及编号Table 1 Sources and number ofOenothera biennis
1.2 仪器与设备
FL 9720气相色谱仪:浙江福立分析仪器有限公司;AUY220型电子分析天平:日本岛津公司;HP-5毛细管气相色谱柱:美国安捷伦科技有限公司;氢火焰离子化检测器:浙江福立分析仪器有限公司;SHC型高纯氢气发生器、JK-5L型纯净空气泵:山东赛克赛斯氢能源有限公司;BJ-100植物粉碎机:上海拜杰实业有限公司;WSP-100恒温水浴锅:上海申生科技有限公司;SHZ-DⅢ循环水真空泵:巩义市予华仪器有限责任公司;SHZ-A水浴恒温振荡器:常州国旺仪器有限公司;H1850台式高速离心机:湖南长沙湘仪离心机厂。
1.3 方法
1.3.1 色谱条件的优化方法
GC9720色谱柱(0.25 mm×30 m×0.25 μm);分流比30∶1;检测器温度280℃;载气高纯氮气;柱流量1.5 mL/min;氢气流量30mL/min;空气流量300 mL/min;进样量2 μL;柱温箱温度:在实验条件优化过程中共筛选了3个升温程序。
A:初始温度50℃,保持2min,以10℃/min升温至100℃,保持2 min,以15℃/min升温至140℃,保持5 min,以8℃/min升温至200℃,保持10 min。
B:初始温度55℃,保持2min,以7℃/min升温至120℃,保持3 min,以16℃/min升温至180℃,保持5 min,以8℃/min升温至220℃,保持12 min。
C:初始温度55℃,保持3min,以5℃/min升温至100℃,保持2 min,以20℃/min升温至160℃,保持3 min,以3℃/min升温至230℃,保持15 min。1.3.2供试品溶液制备方法
取自然晾干的月见草适量,粉碎,过50目筛,准确称量100 g,置于2 000 mL圆底烧瓶中,加正己烷1 200 mL,水浴加热回流提取2次,每次1.5 h,合并提取液,减压回收溶剂,得月见草油。由于正己烷直接萃取的月见草提取液中可能会混有部分长链脂肪酸或不饱和脂肪酸,若直接进气相分析,会污染毛细柱,因此需将在进样前对其进行衍生化,本文采用氢氧化钾-甲醇溶液进行脂肪酸甲酯化的方法进行样品处理,在处理过程中,分别对氢氧化钾-甲醇溶液浓度、加入量、反应时间3个因素进行考察。具体操作为:取月见草油约50 mg,置于具塞离心试管中,加入5 mL正己烷中,然后分别加入浓度为(1 mol/L、2 mol/L、3 mol/L)的KOH甲醇溶液[14](0.5 mL、1.0 mL、2.0 mL),室温振荡反应(2 min、3 min、5 min、10 min),以1 200 r/min的转速进行离心,取上层有机相,置于10 mL容量瓶中,正己烷定容,摇匀,进样前经0.45 μm微孔滤膜过滤。
1.3.3 标准品溶液的制备
分别精密称定γ-亚麻酸甲酯0.003 0 g、亚油酸甲酯0.005 0 g,分别置于两个5 mL棕色容量瓶中,加正己烷溶解并定容至刻度,即得γ-亚麻酸甲酯、亚油酸甲酯标准品贮备液;分别精密移取上述两个标准品贮备液0.4 mL,分别置于同一个5 mL容量瓶中,即得体积分数分别为48 μL/mL和80 μL/mL的γ-亚麻酸和亚油酸混合标准品溶液,冷藏备用。
1.3.4 数据处理
指纹图谱相似度计算采用国家药典委员会“中药色谱指纹图谱相似度评价系统软件”2012.130723版本,计算方法采用均值法;聚类分析采用SPSS20.0软件,以月见草油指纹图谱各共有峰面积为数据源,采用系统聚类中的组间连接方法,样品距离度量采用平方Euclidean距离进行计算;利用SIMCA 14.1软件,以不同批次月见草油各共有峰的峰面积为自变量,进行主成分分析。
2 结果与分析
2.1 条件优化
在柱温箱温度优化过程中因C升温程序的色谱图中,各色谱峰分离度良好,色谱峰分布均匀,故最终选择C升温程序进行实验;供试品溶液甲酯化试验结果表明,在相同的色谱条件下,加入2 mol/L的KOH甲醇溶液1.0 mL,室温振荡反应5 min,色谱峰较多、峰面积较大,且样品处理时间合适,因此在供试品溶液制备过程中,采用向样品溶液中加入2 mol/L的KOH甲醇溶液1.0 mL,室温振荡反应5 min的方法进行甲酯化。将表1中各批月见草药材分别按“1.3.2”项下优化出的最佳方法制备供试品溶液,并按“1.3.1”项优化出的最佳色谱条件测定,记录结果见图1(图中显示样品S4的色谱图)。
2.2 月见草油指纹图谱的建立
2.2.1 参比峰的选择
通过气相色谱法对10批月见草油进行测定,在样品的气相色谱图(图1)中,亚油酸甲酯(4号峰)色谱峰的峰面积较大,与相邻色谱峰的分离度较好,且为10批月见草油指纹图谱所共有,因此,本研究将其设为参照峰,用以计算其他共有峰的相对保留时间和相对峰面积。

图1 月见草油样品S4(A)和混合标准品(B)气相色谱图Fig.1 GC chromatogram ofOenothera biennisoil sample S4(A)and mix standards(B)
2.2.2 指纹图谱的建立及相似度评价
将所得的10批月见草挥发油GC图谱以AIA格式依次导入中国药典委员会研制的中药色谱指纹图谱相似度评价系统(2012.130723版),建立了月见草挥发油GC指纹图谱叠加模式色谱图,结果如图2所示。将10批月见草油按平均值法生成对照指纹图谱,通过对各批次的指纹图谱的相似度计算,结果见表2。

图2 10批月见草油GC指纹图谱叠加图Fig.2 GC fingerprint superposition of 10 batches of Oenothera biennisoil
由图2可知,以样品S4药材图谱作为参照谱进行指纹匹配,确定了8个共有峰。由表2可知,以10批月见草油按平均值法生成对照指纹图谱相似度为1进行计算,10批药材的相似度在0.914~0.989范围内。
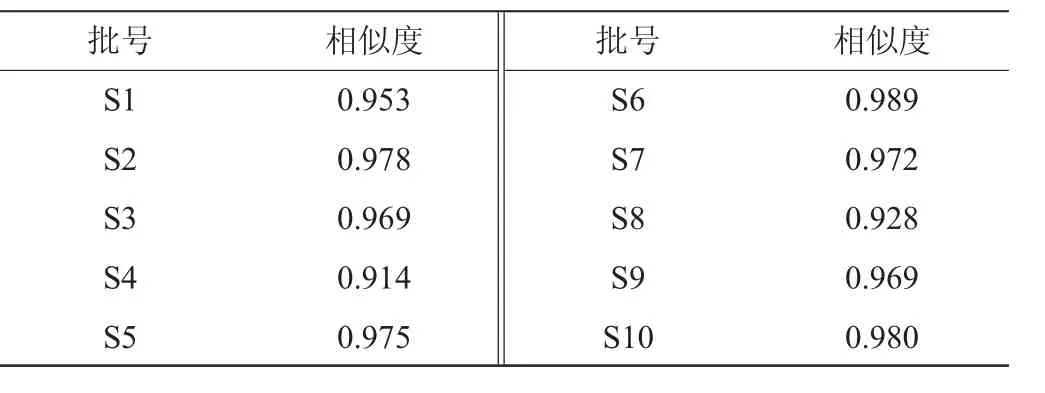
表2 10批月见草挥发油GC指纹图谱相似度Table 2 Similarity of GC fingerprint of the 10 batches of Oenothera biennisoil
2.3 方法学考察
2.3.1 精密度试验
取同一供试品溶液(样品S4),按“1.3.1”项下方法优化出的最佳色谱条件,连续进样5次,记录色谱图,以4号峰(亚油酸甲酯)为参照峰,计算各共有指纹峰的相对保留时间和相对峰面积结果相对标准偏差(relative standard deviation,RSD)分别见表3和表4。
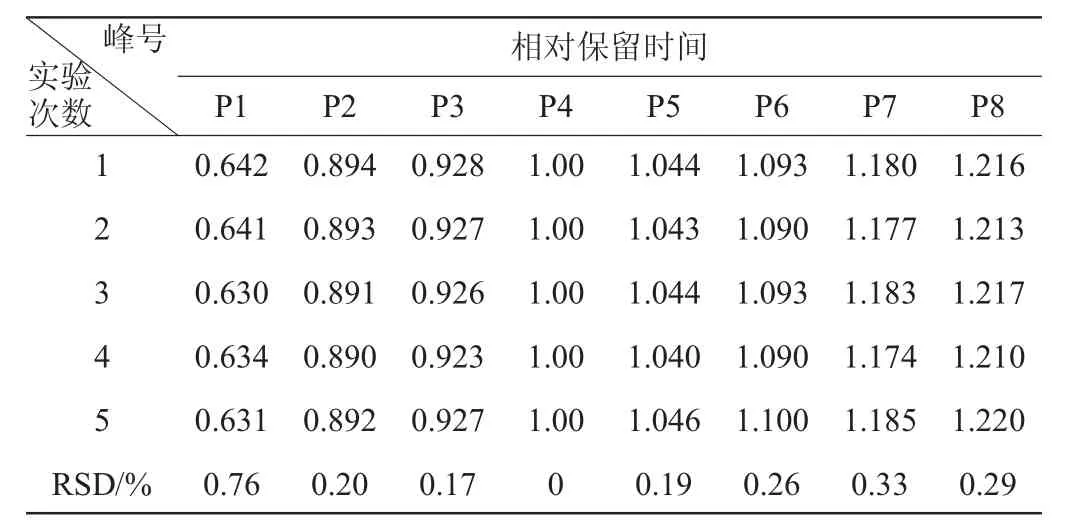
表3 相对保留时间的精密度试验结果Table 3 Experimental results of precision for relative retention time

表4 相对峰面积的精密度试验结果Table 4 Experimental results of precision for relative peak area
由表3可知,各共有峰相对保留时间的相对标准偏差RSD<0.76%;由表4可知,相对峰面积的RSD<1.60%,表明方法的精密度良好。
2.3.2 稳定性试验
取同一供试品(样品S5)溶液,按“1.3.1”项下方法优化出的最佳色谱条件,分别于2 h、4 h、6 h、8 h、12 h进样,记录色谱图,以4号峰(亚油酸甲酯)为参照峰,计算各共有指纹峰的相对保留时间和相对峰面积见表5和表6。
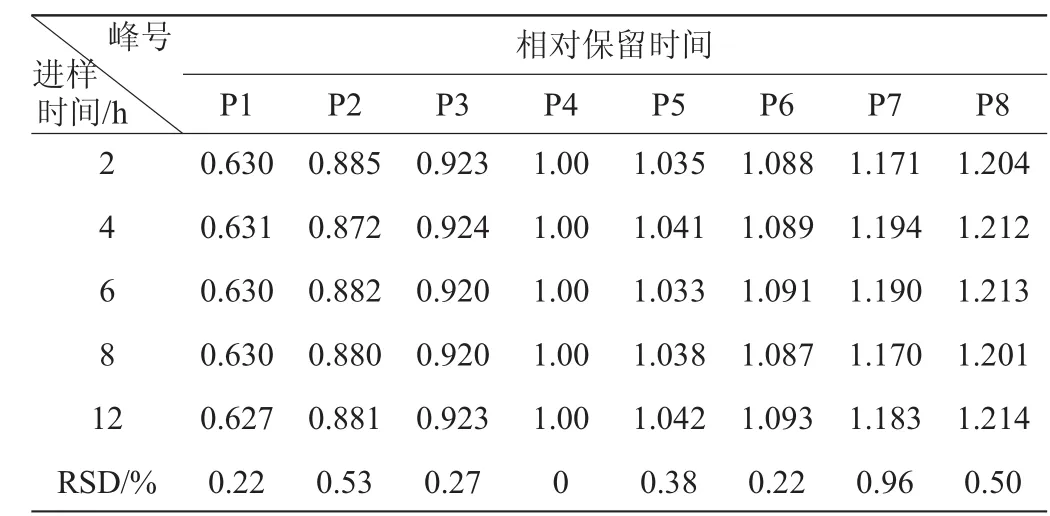
表5 相对保留时间的稳定性试验结果Table 5 Experimental results of stability for relative retention time

表6 相对峰面积的稳定性试验结果Table 6 Experimental results of stability for relative peak area
由表5、表6可知,各共有峰相对保留时间RSD<0.96%,各共有峰相对峰面积RSD<1.85%。表明供试品溶液在12 h内稳定。
2.3.3 重复性试验
取同一批(样品S4)月见草药材5份,按“1.3.2”项下方法优化出的最佳供试品溶液制备方法平行制备5份月见草油供试品溶液,按“1.3.1”项下方法优化出的最佳色谱条件,依次测定,记录色谱图,以4号峰(亚油酸甲酯)为参照峰,计算各共有指纹峰的相对保留时间和相对峰面积见表7和表8。
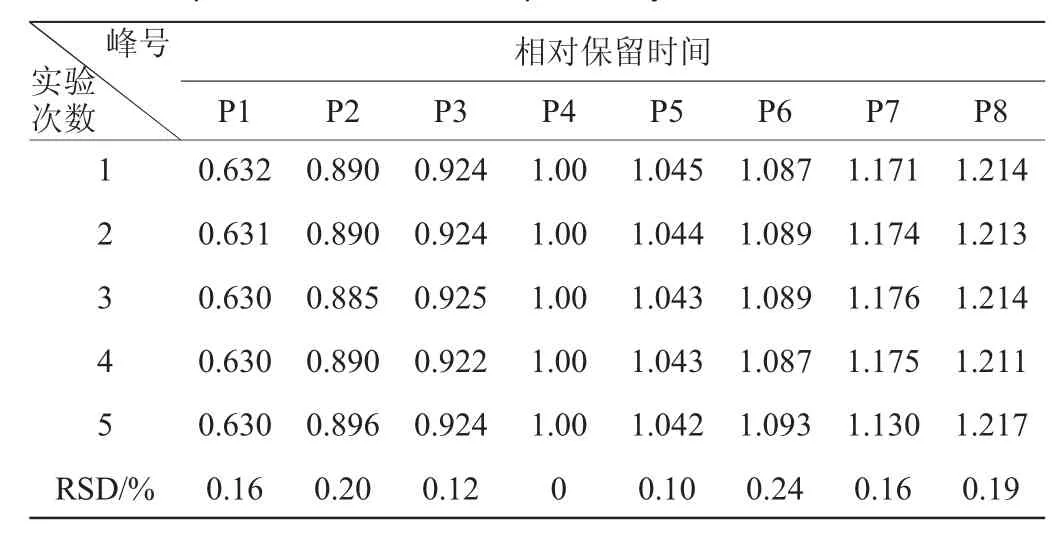
表7 相对保留时间的重复性试验结果Table 7 Experimental results of repeatability for relative retention time

表8 相对峰面积的重复性试验结果Table 8 Experimental results of repeatability for relative peak area
由表7、表8可知,各共有峰相对保留时间RSD<0.24%,各共有峰相对峰面积RSD<2.83%,表明该方法具有较好的重复性。
2.4 月见草油指纹图谱的聚类分析
本研究以月见草油指纹图谱各共有峰面积为数据源[15],对10批月见草油的GC指纹图谱进行聚类分析,采用系统聚类中的组间连接方法,样品距离度量采用平方Euclidean距离,得到10批月见草油指纹图谱的聚类分析树状图,结果如图3所示。
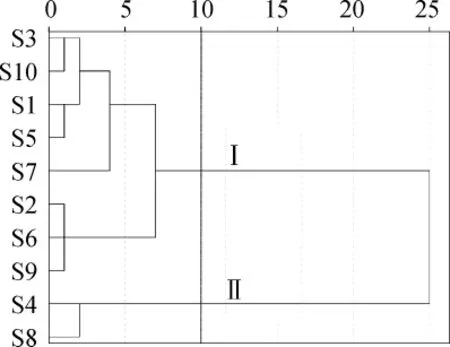
图3 10批月见草油指纹图谱的聚类分析树状图Fig.3 Cluster analysis tree for 10 batches ofOenothera biennisoil
聚类分析图反映的是各样品的相似程度,样品间的分类距离值越小则样品间的差异越小,反之则越大。由图3可知,当分类距离取10时,10批月见草样品被分为2类,即Ⅰ和Ⅱ类,其中S4和S8为II类,其余样品为I类。
2.5 主成分分析
以主成分特征值>1作为选择主成分的依据,获得2个主成分,主成分分析结果如图4所示。
由图4可知,累计均方差为96.5%,表明引入的2个主成分可以表达96.5%的原变量信息,从图4A中可以看出,10批样本大致分为两组,分别标记为I和II。其中,S4和S8为II组,其余样品为I组,由于S4和S8样品来源分别为湖北巴东和江西吉水,而其余样品均来自北方寒冷地区,结合分析结果,地理位置与气候条件的差异对月见草质量存在一定程度的影响。从图4B中可以看出1,2,3,4,5,6号峰对PC1有较大的贡献,而7,8号峰对PC2有较大的贡献。
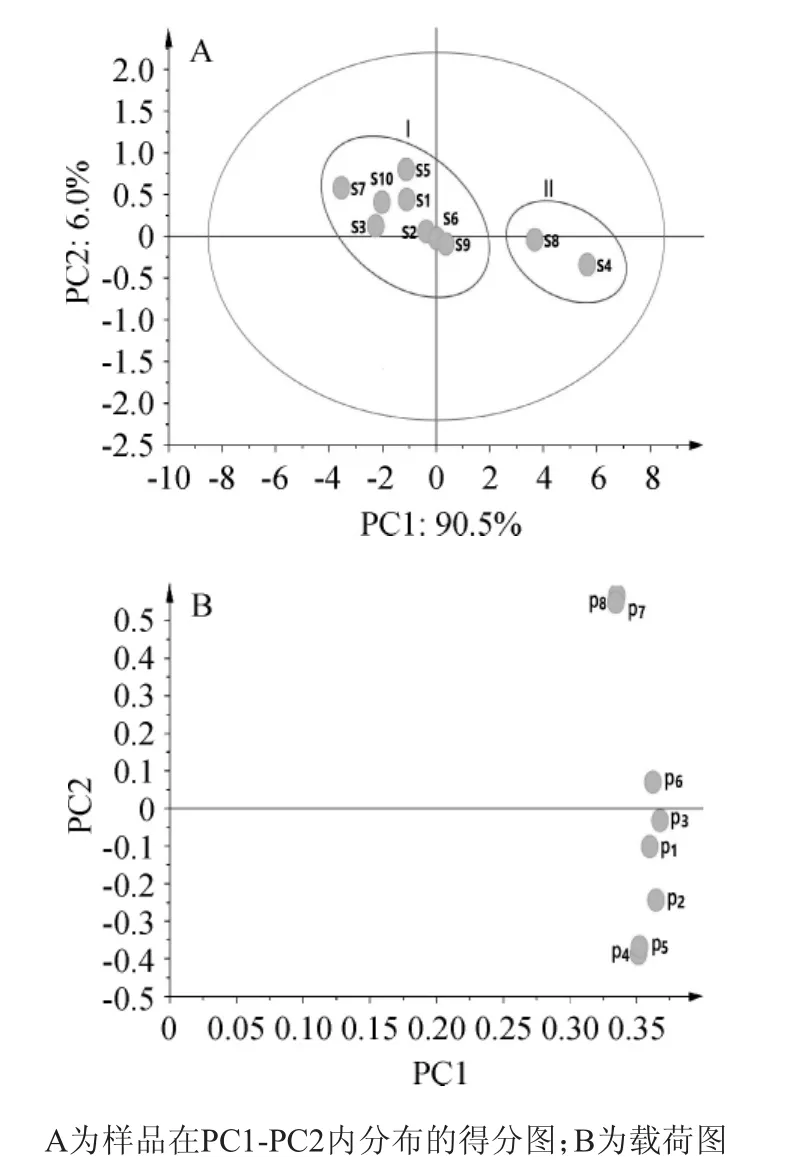
图4 不同批次月见草油指纹图谱的主成分分析结果Fig.4 Results of principle components analysis of fingerprint of oil fromOenothera bienniswith different batches
3 结论
在最优的样品处理方法和色谱条件下,建立了月见草油的指纹图谱,经方法学考察,该方法精密度、重复性和稳定性良好,均符合指纹图谱规定要求。通过对指纹图谱分析,确证了8个共有峰,10批月见草油的相似度为0.914~0.989。考虑到中药这一复杂体系中的不同的化学物质之间可能存在着一定的关联性,因此聚类分析和主成分分析被应用于此研究中,并且聚类分析和主成分分析的结果一致,所选样本在两种方法下均被分成了两类,同时主成分分析结果表明1、2、3、4、5、6号峰为影响月见草油指纹图谱相似度评价的主要因素(累计方差为90.5%)。由于受实验条件所限,本实验只鉴定出4号峰和5号峰分别为亚油酸甲酯和γ-亚麻酸甲酯,其它色谱峰代表的化学成分需进一步研究。本研究建立的月见草油指纹图谱结合化学计量学的方法,为月见草油质量控制提供了依据。
猜你喜欢
西北药学杂志(2023年4期)2023-07-27 07:19:14
中学生百科·大语文(2023年2期)2023-05-08 09:27:17
中医药导报(2021年4期)2021-11-22 12:21:12
云南教育·中学教师(2019年5期)2019-08-13 07:02:38
中国洗涤用品工业(2019年4期)2019-05-11 09:27:18
世界科学技术-中医药现代化(2018年9期)2019-01-29 03:40:50
儿童时代·快乐苗苗(2016年5期)2016-09-14 22:47:12
食品界(2016年4期)2016-02-27 07:37:06
中国粮油学报(2015年5期)2015-02-06 01:47:28
中国刑警学院学报(2014年4期)2014-04-27 10:08:00
