
Mitochondrial division inhibitor 1 protects cortical neurons from excitotoxicity: a mechanistic pathway
2018-08-09 01:27:24KuaiZhouHaiYuanYangPengYuTangWeiLiuYongJunLuoBinLvJianYinTaoJiangJianChenWeiHuaCaiJinFan
中国神经再生研究(英文版) 2018年9期
Kuai Zhou , Hai-Yuan Yang , Peng-Yu Tang Wei Liu Yong-Jun Luo Bin Lv Jian Yin, Tao Jiang Jian Chen Wei-Hua Cai , Jin Fan
1 Department of Orthopedics, First Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu Province, China
2 Department of Orthopedics, BenQ Hospital Affiliated to Nanjing Medical University, Nanjing, Jiangsu Province, China
3 Department of Orthopedics, Affiliated Jiangning Hospital of Nanjing Medical University, Nanjing, Jiangsu Province, China
Abstract Mitochondrial division inhibitor 1 (Mdivi-1) is a selective cell-permeable inhibitor of dynamin-related protein-1 (Drp1) and mitochondrial division. To investigate the effect of Mdivi-1 on cells treated with glutamate, cerebral cortex neurons isolated from neonatal rats were treated with 10 mM glutamate for 24 hours. Normal cultured cells and dimethyl sulfoxide-cultured cells were considered as controls. Apoptotic cells were detected by flow cytometry. Changes in mitochondrial morphology were examined by electron microscopy. Drp1, Bax, and caspase-3 expression was evaluated by western blot assays and immunocytochemistry. Mitochondrial membrane potential was detected using the JC-1 probe.Twenty-four hours after 10 mM glutamate treatment, Drp1, Bax and caspase-3 expression was upregulated, Drp1 and Bax were translocated to mitochondria, mitochondrial membrane potential was decreased and the rate of apoptosis was increased. These effects were inhibited by treatment with 50 μM Mdivi-1 for 2 hours. This finding indicates that Mdivi-1 is a candidate neuroprotective drug that can potentially mitigate agai nst neuronal injury caused by glutamate-induced excitotoxicity.
Key Words: nerve regeneration; mitochondrial division inhibitor 1; neurons; apoptosis; mitochondria; division; dynamin-related protein-1;phospho-dynamin-related protein-1; Bax; glutamate; colocalization; neural regeneration
Introduction
Glutamate is one of the most important amino acids in the mammalian brain. Glutamate participates in information transmission and energy metabolism and plays an important role in the development of neurological abnormalities, including acute craniocerebral and spinal cord injury, epilepsy, and Parkinson’s and motor neuron diseases. Neuronal depolarization leads to the release of glutamate during neurotransmission, which ceases with glutamate reuptake. The persistence of excessively high concentrations of glutamate in the synaptic cleft can cause neuronal damage and death, a phenomenon known as excitotoxicity (Villacé et al., 2017).
Mitochondria in eukaryotic cells function as the main sites of adenosine triphosphate production by mitochondrial oxidative phosphorylation. Mitochondrial dysfunction in the nervous system is strongly associated with acute or chronic injury. High concentrations of glutamate binding to receptors on the postsynaptic neuronal membrane stimulate a massive influx of Ca2+, leading to mitochondrial Ca2+overload, inhibition of adenosine triphosphate production, and neuronal death. Glutamate-mediated excitotoxicity mainly occurs via the mitochondrial apoptosis pathway (Xu et al., 2017). This is associated with the production of reactive oxygen species that block mitochondrial oxidative phosphorylation and increase mitochondrial membrane permeability, resulting in the release of cytochrome c (cyto c) and activation of the caspase cascade(Jia et al., 2016).
A balance is maintained between mitochondrial division and fusion (Oettinghaus et al., 2016; Kim et al., 2017a), which is important for normal mitochondrial form and function (Miwa and Saretzki, 2017). Dynamin-related protein 1 (Drp1), mitochondrial fission protein 1 (Fis1), and mitochondrial fission factor (MFF) regulate mitochondrial division, while mitofusin 1/2 (Mfn1/2) and optic atrophy 1 (Opa1) are involved in mitochondrial fusion. Both processes are, however, linked to cell apoptosis. Therapeutic strategies that restore normal mitochondrial structure and reduce excessive division can potentially be useful for the treatment of nerve injury (Xu et al.,2016).
Drp1 overexpression increases apoptosis in pancreatic beta cells induced by high sugar (Reinhardt et al., 2016), while excessive division of mitochondria in nerve cells caused by Drp1 overexpression has been linked to Huntington’s disease(Reddy, 2014). Mitochondrial division inhibitor 1 (Mdivi-1)is a Drp1 inhibitor that blocks mitochondrial division (Reddy,2014) and thereby decreases apoptosis of hippocampal (Agarwal et al., 2016) and spinal cord (Liu et al., 2015) neurons.However, the mechanism by which Drp1 modulates neuronal injury induced by glutamate remains unclear. In this study, we investigated the effect of Mdivi-1 on mitochondrial apoptosis using a cortical neuron culture model of glutamate-induced excitotoxicity.
Materials and Methods
Cell culture
Thirty specific-pathogen-free, male, neonatal (24-hour-old)Sprague-Dawley rats were obtained from Nanjing University,China (license No. SCXK2015-0001). This study was approved by the Institutional Animal Care and Use Committee of Nanjing Medical University, China (approval No. 1601036).Neonatal rats were placed in 75% ethyl alcohol for 1 minute and then sacrificed. Rats were decapitated and heads put in Dulbecco’s Modified Eagle’s Medium (DMEM)/F12(HyClone, Logan, UT, USA) on ice. The endocranium was cut along the midline and the brain removed. The pia mater was wiped off, the brain cut into pieces (1 mm × 1 mm × 1 mm) and then digested with pancreatic enzymes (0.25%) and DNase I for 20 minutes at 37°C. The digestion was terminated by the addition of horse serum (Gibco, Logan, UT, USA).The medium containing neurons was centrifuged at 55.95 × g for 5 minutes at 4°C. Isolated neurons were then seeded in 6-or 24-well plates in DMEM/F12 supplemented with 10% horse serum, 100 U/mL penicillin (Gibco), 100 μg/mL streptomycin(Gibco), and 1% glutamine and cultured for 4 hours in a humidified incubator at 37°C and 5% CO2. The medium was then changed to Neurobasal A (Gibco) supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin, and 1% glutamine (Gibco).
Establishment of glutamate-induced neuronal injury models
Neurons isolated from neonatal rats were randomly divided into four groups: control, dimethyl sulfoxide, glutamate, and Mdivi-1 + glutamate. Cells in the control group were left untreated. In the dimethyl sulfoxide group, cells were cultured in medium containing dimethyl sulfoxide (the ratio of dimethyl sulfoxide to medium was 1:200; Sigma). Cells in the glutamate and Mdivi-1 + glutamate groups were treated with 10 mM L-glutamic acid (Sigma, Shanghai, China) for 24 hours (Zhu et al., 2017); cells in the Mdivi-1 + glutamate group were pretreated with 50 μM Mdivi-1 (Sigma) for 2 hours before addition of L-glutamic acid.
Detection of apoptosis by flow cytometry
The Annexin V-FITC Apoptosis Detection kit (Invitrogen,Guangzhou, China) was used to prepare samples for flow cytometry. Cells were digested with trypsin (Gibco) without ethylenediamine tetraacetic acid (Gibco), then washed in phosphate-buffered saline (PBS) and centrifuged twice at 223.8 × g for 5 minutes. The pellet was resuspended in 500 μL binding buffer (5 × 105cells) and then mixed with 5 μL Annexin V-FITC and 5 μL propidium iodide. After incubation in the dark at room temperature for 10 minutes, cells were sorted over the course of 1 hour. Cells in the left lower quadrant were normal, while cells in the right lower quadrant were apoptotic.
Detection of mitochondrial membrane potential
JC-1 staining buffer (5×) (Beyotime Institute of Biotechnology, Shanghai, China) was diluted 4-fold in culture medium and placed on ice. Cells were washed in PBS and then incubated in the diluted JC-1 buffer for 20 minutes at 37°C. After two washes in PBS, cells were observed with an epifluorescence microscope (Zeiss, Nanjing, China).
Western blot assays
Cytoplasmic or mitochondrial proteins (30 μg) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane(Bio-Sharp Solutions, Miami, FL, USA). Membranes were blocked with 5% non-fat milk containing 0.1% Tween-20 in Tris-buffered saline at room temperature for 1 hour, and incubated overnight at 4°C with primary antibodies against the following: Bax, p-Drp1, Drp1, cyto c, caspase-3, GAPDH and COX IV (1:1000; mouse or rabbit, monoclonal; Abcam,Shanghai, China), then with horseradish peroxidase-conjugated secondary antibody (1:2500; mouse or rabbit, monoclonal;Abcam) on the following day at room temperature. Protein bands were detected by enhanced chemiluminescence (Thermo Scientific, USA). The optical density of a band represents its protein content (ImageJ, Apache, USA). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and cyto c oxidase subunit IV (COX IV) were used as loading controls.
Immunofluorescence analysis

Figure 1 Expression of microtubule-associated protein 2 (MAP2) in cells isolated from neonatal rats and cultured for 7 days.

Figure 2 Effect of mitochondrial division inhibitor 1 (Mdivi-1) on cell apoptosis ratios of cortical neurons treated with glutamate detected by flow cytometry.

Figure 3 Effect of mitochondrial division inhibitor 1 (Mdivi-1) on the mitochondrial structure of cortical neurons treated with glutamate(transmission electron microscopy, upper,magnification of 1 × 104; lower, magnification of 3 × 104).
Cells were washed with PBS for 5 minutes three times and fixed with 4% paraformaldehyde for 15 minutes before permeabilization with 0.1% Triton X-100 for 20 minutes. Non-specific binding was blocked by incubation in 10% goat serum for 1 hour at room temperature. Samples were then incubated with primary antibodies against the following: microtubule-associated protein 2 (MAP2), COX IV, Drp1 (1:500; mouse or rabbit, monoclonal; Abcam) for 14 hours at 4°C, and then washed three times with PBS for 10 minutes. After incubation with secondary antibody (1:2,000; Cy3- or FITC-mouse or rabbit, monoclonal; Abcam) for 1 hour at room temperature,cells were washed in PBS for 10 minutes three times, stained with DAPI, and visualized under an epifluorescence microscope. Fluorescence intensity increased when the expression of proteins increased. Pictures were merged and used to show the colocalization between Bax and Drp1 in mitochondria.
Transmission electron microscopy
Samples were washed three times in precooled PBS for 10 minutes, digested with 0.25 mM pancreatin for 5 minutes at 37°C, washed in PBS and centrifuged at 55.95 × g and 4°C for 10 minutes. After fixation in 2.5% glutaraldehyde, samples were prepared for transmission electron microscopy according to standard protocols.
Statistical analysis
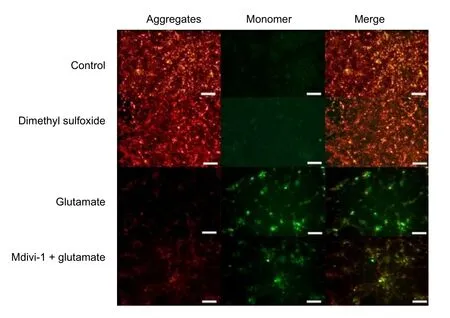
Figure 4 Effect of mitochondrial division inhibitor 1 (Mdivi-1) on the mitochondrial membrane potential of cortical neurons treated with glutamate.

Figure 5 Effect of mitochondrial division inhibitor 1 (Mdivi-1) on Bax, p-Drp1, Drp1, cytochrome c (cyto c), and caspase-3 protein levels in cortical neurons treated with glutamate.
Data are expressed as the mean ± SD and were analyzed using SPSS v.17.0 software (SPSS, Chicago, IL, USA). Experiments were repeated at least three times. Statistical significance was evaluated by one-way analysis of variance. The Independent samples t-test was used for comparison between two groups. A value of P < 0.05 was considered statistically significant.
Results
Identification of neurons
MAP2 is specific to neurons and promotes microtubule network assembly and stability; as such, it is considered as a marker for mature neurons (Ribeiro and de Wit, 2017). Cells isolated from neonatal rats exhibited a neuronal morphology consisting of cell bodies, dendrites, and axons, and expressed MAP2 (Figure 1).
Glutamate increased the apoptosis ratio, which was reversed by Mdivi-1
Flow cytometry revealed that apoptosis ratios were significantly higher in glutamate and Mdivi-1 + glutamate groups than in the control group (P < 0.01; Figure 2).
Mdivi-1 prevented glutamate-induced mitochondrial damage
Neurons cultured for 7 days were observed by transmission electron microscopy. Mitochondrial and nuclear morphology was normal in control and dimethyl sulfoxide groups; mitochondria were round, oval, star, or rod-shaped. However, in the glutamate group, a few mitochondria had a normal shape,many appeared swollen, with a ruptured mitochondrial outer membrane. Glutamate toxicity led to pyknosis and apoptosis.In the Mdivi-1 + glutamate group, there was less damage to mitochondria induced by glutamate; the outer membrane was relatively intact in most cases, and mitochondria retained a round or oval form (Figure 3).
Reduction in mitochondrial membrane potential induced by glutamate was blocked by Mdivi-1
JC-1, a fluorescent probe, is widely used for the detection of mitochondrial membrane potential (Contino et al., 2017).JC-1 aggregates in the mitochondrial matrix and J-aggregates show red fluorescence when the mitochondrial membrane potential is relatively high. However, JC-1 does not aggregate in the mitochondrial matrix and the JC-1 monomer produces green fluorescence when the mitochondrial membrane potential is low. There was no difference between green and red fluorescence intensities in control and dimethyl sulfoxide groups.However, the intensity of green fluorescence was enhanced relative to that of red fluorescence in the Mdivi-1 + glutamate group and especially in the glutamate group. This indicated that glutamate can reduce the mitochondrial membrane potential and that this effect can be mitigated by pretreating with Mdivi-1 (Figure 4).
Glutamate induced dephosphorylation of p-Drp1 and Drp1 translocation to mitochondria
Drp1 exists in the cytoplasm in a phosphorylated form (Ser637)and regulates mitochondrial division. Increased cytoplasmic Ca2+concentration activates calcium-induced neurotransmission, resulting in the dephosphorylation of p-Drp1 (Perdiz et al., 2017). Hyperactivation of Drp1 increases mitochondrial division. P-Drp1 in cytoplasmic and mitochondrial extracts was detected by western blot assay (Figure 5). In cells treated with glutamate, the mitochondrial p-Drp1 level was decreased whereas that of Drp1 was increased, an effect that was abolished by Mdivi-1. The expression level and cellular distribution of Drp1 were also examined by immunocytochemistry (Figure 6). The mitochondrial marker COX IV was used to identify mitochondria. There was no difference in Drp1 expression level or distribution between control and dimethyl sulfoxide groups; the Drp1 expression level was higher in the Mdivi-1 +glutamate group and especially in the glutamate group compared with the controls. Glutamate increased the colocalization of Drp1 and COX IV, an effect that was abolished by Mdivi-1.
Glutamate increased Bax activation and translocation to mitochondria
Bax is a pro-apoptotic Bcl-2 family member that induces mitochondrial apoptosis. Bax also mediates the decrease in mitochondrial membrane potential caused by accumulation of reactive oxygen species (Hu and Xiao, 2015). Bax levels in total cell extracts were increased in the Mdivi-1 + glutamate group and especially in the glutamate group (Figure 5). The combined Bax levels in cytoplasm and mitochondria were similar to Bax levels in whole cell extracts.
Glutamate stimulated cyto c release from mitochondria to cytoplasm
In the cytoplasm of glutamate and Mdivi-1 + glutamate groups, cyto c levels were lower than in the control group (Figure 5). There was no difference in cyto c levels in total cell extracts of the four groups; however, the groups differed in terms of cyto c distribution in the cytoplasm and mitochondria: the level in the cytoplasm was higher and in mitochondria lower in the Mdivi-1 + glutamate group and especially in the glutamate group compared with the control group (P < 0.01).
Glutamate increased cytoplasmic caspase-3
The cytoplasmic caspase-3 level in the Mdivi-1 + glutamate group and especially in the glutamate group was higher than that in the control group (P < 0.01; Figure 5). Glutamate induced apoptosis by increasing cytoplasmic caspase-3 levels, an effect that was abolished by Mdivi-1.
Glutamate increased levels and mitochondrial localization of Bax
The distribution of Bax was similar to that of Drp1 in total cell,cytoplasmic, and mitochondrial extracts (Figure 5). Glutamate increased Bax levels (Figure 6), as determined by immunofluorescence; there was no difference in Bax level between control and dimethyl sulfoxide groups, but the level in the glutamate and Mdivi-1 + glutamate groups was higher than that in the control group. Glutamate increased Bax and COX IV colocalization, an effect that was inhibited by Mdivi-1 (Figure 7).
Drp1 and Bax were colocalized in mitochondria in the presence of glutamate
Drp1 and Bax were upregulated and transported to mitochondria in cells treated with glutamate (Figure 5). To determine whether Drp1 and Bax interact in mitochondria, we analyzed colocalization of the two proteins by immunocytochemistry(Figure 8). Glutamate increased Drp1 and Bax colocalization in mitochondria, which was blocked by Mdivi-1 (Figure 8).
Discussion
Neuronal injury induced by glutamate was associated with mitochondria
The mechanism of glutamate-induced neuronal injury is still not clear (Shi et al., 2016). It is widely accepted that glutamate binds to its receptor on the postsynaptic membrane and activates Ca2+channels, leading to cytoplasmic and mitochondrial Ca2+overload (Wu et al., 2017). This can in turn activate Ca2+-dependent enzymes that degrade phospholipids, DNA,and proteins. An increase in the production of reactive oxygen species can also undermine mitochondrial functioning by blocking the respiratory chain, leading to cell death (Park and Jonas, 2014). We observed that glutamate treatment increased cyto c release and the apoptosis ratio of cells. It also destroyed normal mitochondrial structure, which decreased mitochondrial membrane potential and increased permeability; the resultant release of cyto c and caspase-3 activation likely induced apoptosis. We also found that the pro-apoptotic protein, Bax,could destroy mitochondrial function and this effect could be inhibited by Mdivi-1, a Drp1 inhibitor. Maintaining the homeostasis of mitochondria can inhibit apoptosis induced by glutamate and glutamate induced apoptosis by promoting the colocalization between Bax and Drp1 in mitochondria.
Glutamate induced neuronal apoptosis by promoting the dephosphorylation of p-Drp1 and this effect could be inhibited by Mdivi-1
The balance between mitochondrial division and fusion is essential for normal functioning; perturbation of this balance can lead to apoptosis or necrosis (Wang et al., 2014). Mitochondrial fusion is regulated by Mfn1, Mfn2, and Opa1 and requires the activity of GTPase (Wang et al., 2014). In mammals, Drp1 and Fis1 regulate mitochondrial division. Unlike Fis1, Drp1 lacks a transmembrane domain that is required for binding to the mitochondrial membrane, and exists in the cytoplasm in a phosphorylated form. When cells are stimulated, Fis1, which has a transmembrane domain, may mediate the translocation of Drp1 from the cytoplasm to mitochondria so that mitochondrial division can proceed (Fu et al., 2017) .
Mdivi-1 decreases neuronal damage induced by glutamate and restores mitochondrial membrane potential decreased by reactive oxygen species (Wu et al., 2017), thus preventing adriamycin-induced myocardial injury. Treatment of hippocampal neurons with jaborandi alkali can result in excessive mitochondrial division and cyto c release, which is inhibited by Mdivi-1(Xie et al., 2016). In this study, mitochondrial membrane potential was decreased by treatment with glutamate, an effect that was reversed by Mdivi-1. Our transmission electron microscopy analysis revealed that glutamate caused mitochondrial swelling and vacuolization and nuclear pyknosis in neurons,which was abrogated by pretreatment with Mdivi-1. Glutamate decreased the level of cytoplasmic p-Drp1 and increased mitochondrial Drp1 by inducing p-Drp1 dephosphorylation and the translocation of Drp1 from the cytoplasm to mitochondria,resulting in activation of apoptosis.
Mdivi-1 blocked neuronal apoptosis by preventing mitochondrial colocalization of Drp1 and Bax
Drp1 is a key mitochondrial shaping protein that is essential for mitochondrial fragmentation and cristae remodeling. Drp1 ablation inhibited activation of the mitochondrial apoptosis pathway induced by Bax and oxidative damage (Oettinghaus et al., 2016). Bax is a proapoptotic molecule that can activate the mitochondrial domain, increase permeability of the outer mitochondrial membrane and lead to the loss of integrity of mitochondrial structure and function (Park and Jonas, 2014).Cells without Drp1 are resistant to apoptotic inducers and oxidative stress (Zhao et al., 2017). Drp1-controlled mitochondrial permeability associated with cyto c release and calcium overload and downstream activation of caspase is facilitated by Bax (Wang et al., 2015). Taken together, these findings indicate that Drp1 collaborates with Bax and regulates apoptosis.Bax is a proapoptotic cytoplasmic protein that is activated in cells treated with glutamate (Kim et al., 2017b). Glutamate promotes Bax/Bax oligomerization and the translocation of Bax to mitochondria (Kim et al., 2017c). Drp1 mutation does not affect Bax localization in mitochondria, but blocks its activation,whereas Drp1 overexpression induces translocation of Bax to mitochondria and its activation, thereby stimulating mitochondrial division. Drp1 does not directly bind mitochondria because it lacks a transmembrane domain (Clinton and Mears,2017). Proapoptotic factors activate mitochondrial division and promote mitochondrial localization of Drp1, giving rise to the possibility that another protein mediates the interaction of Drp1 with mitochondria. The observed colocalization of Drp1 and Bax suggests the latter as a candidate adapter. Mdivi-1 inhibited this colocalization and suppressed apoptosis, although the mechanistic basis for this effect remains to be determined.Spinal cord injury consists of two phases: an initial trauma and a secondary injury of progressive apoptotic damage(Jia et al., 2016). During the secondary injury, mitochondrial membrane potential is reduced and the integrity of the outer membrane is broken, leading to calcium overload, the release of cyto c, and oxidative stress. Balanced mitochondrial homeostasis is essential for axon regeneration. Protection of mitochondrial respiration may promote the recovery of spinal cord function. Maintaining the homeostasis and function of mitochondria may be a potential treatment strategy for spinal cord injury.
The Drp1/Bax/cyto c/caspase-3 pathway mediated glutamate-induced mitochondrial apoptosis
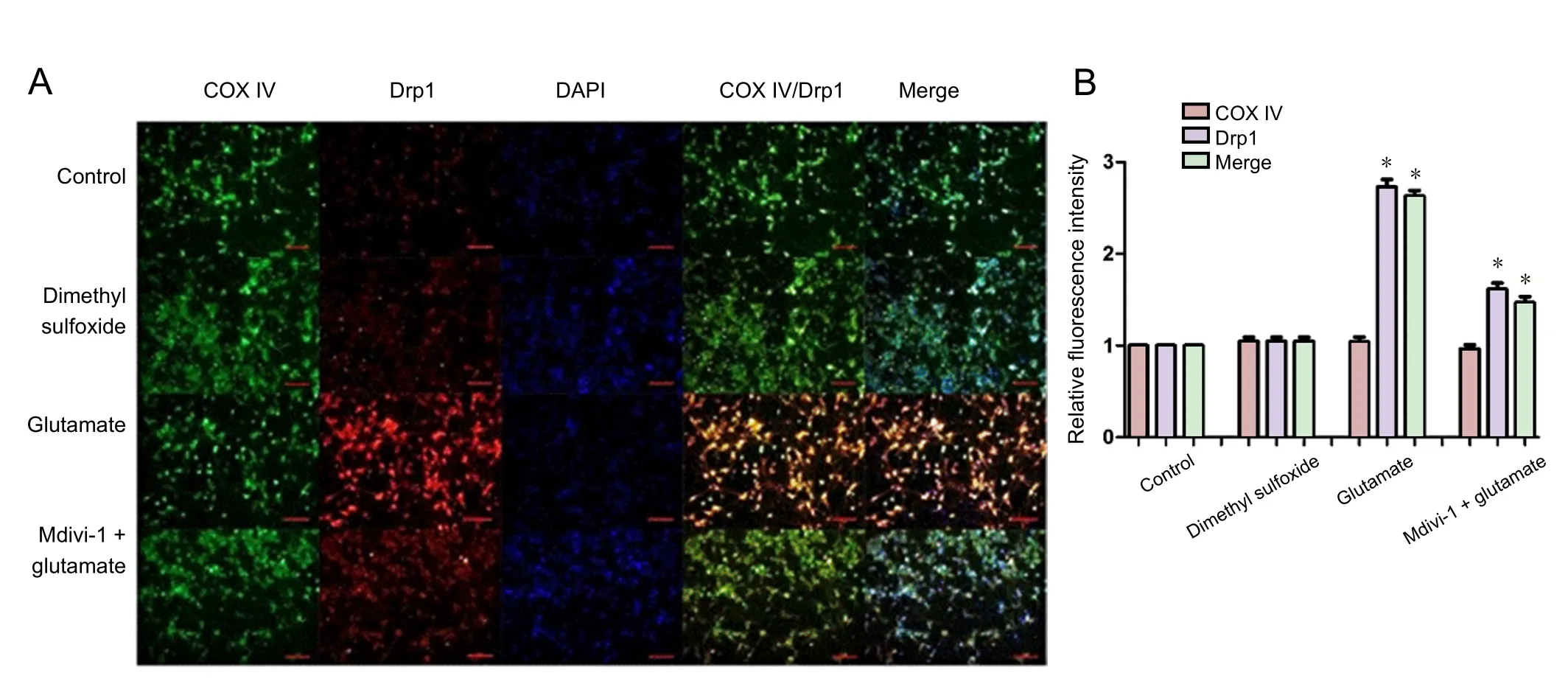
Figure 6 Effect of mitochondrial division inhibitor 1 (Mdivi-1) on expression and subcellular distribution of Drp1 in cortical neurons treated with glutamate.

Figure 7 Effect of mitochondrial division inhibitor 1 (Mdivi-1) on the expression and subcellular distribution of Bax in cortical neurons treated with glutamate.
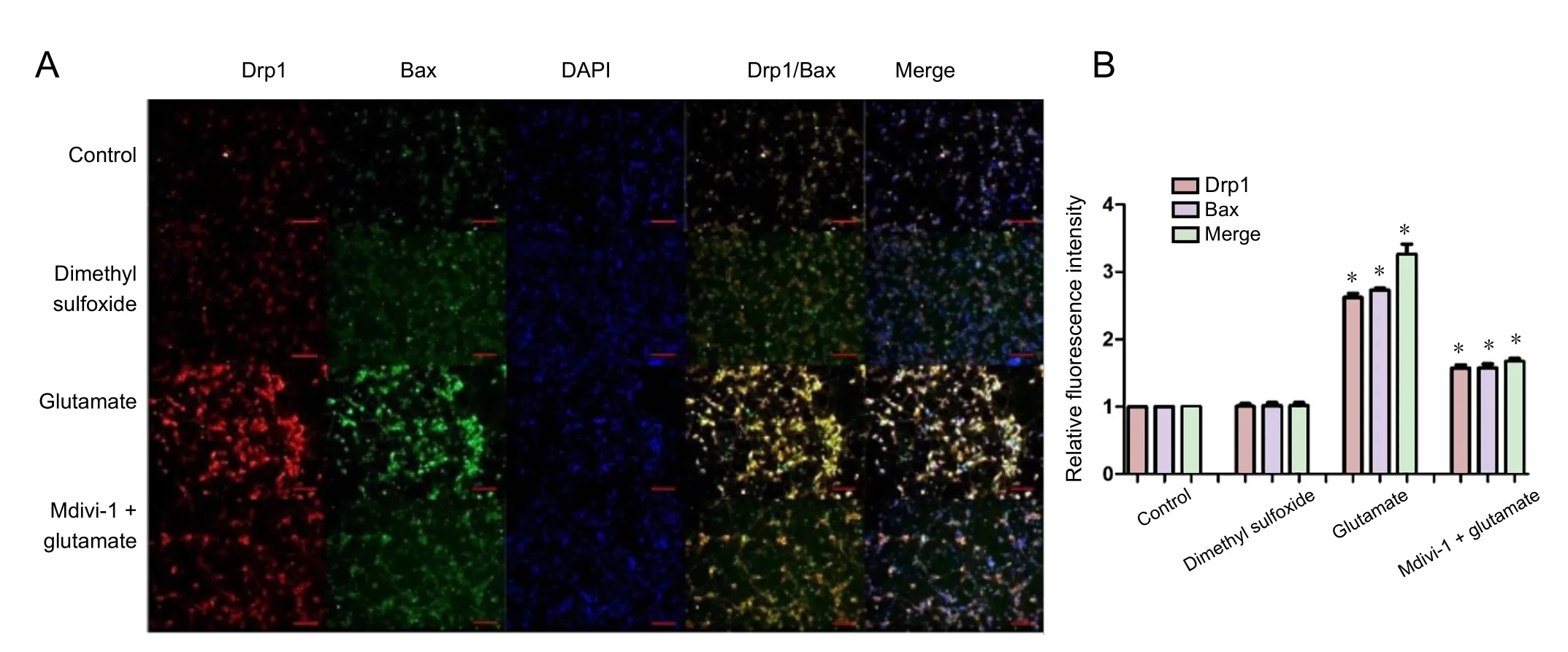
Figure 8 Effect of mitochondrial division inhibitor 1 (Mdivi-1) on expression and mitochondrial colocalization of Drp1 and Bax in cortical neurons treated with glutamate.
Mdivi-1 prevents ischemia-reperfusion injury in rat neurons by inhibiting the release of cyto c, blocking mitochondrial division, and preventing caspase-3 activation. In a spinal cord injury model, Mdivi-1 prevented the decrease in mitochondrial membrane potential and the release of mitochondrial apoptosis-inducing factor. The increases in apoptosis rate and cytoplasmic cyto c and caspase-3 levels induced by treatment with glutamate were abrogated by Mdivi-1. Thus, glutamate may induce apoptosis via a Drp1/Bax/cyto c/caspase-3 signaling cascade that increases mitochondrial membrane permeability,cyto c release, and caspase-3 activation. We found that Bax and Drp1 colocalized in mitochondria and induced apoptosis;however, whether Drp1 colocalized with other pro-apoptosis proteins needs further investigation. Bax and Drp1 colocalization in mitochondria may be an important target for anti-apoptosis and neuronal protection.
Conclusions
The neurotoxicity of glutamate is associated with changes in mitochondrial structure and function. Glutamate increased Drp1 expression, which induced Bax activation; this in turn caused the translocation of Drp1 from the cytoplasm to mitochondria where it acted in conjunction with Bax to decrease mitochondrial membrane potential and increase mitochondrial division and permeability. The latter caused the release of cyto c, which activated caspase-3, leading to apoptosis.Mdivi-1 inhibited Drp1 expression and its translocation to mitochondria, blocking the activation of Bax and caspase-3 and mitochondrial division. By preserving mitochondrial structure and function, Mdivi-1 prevented the decrease in mitochondrial membrane potential and cyto c release caused by glutamate,and consequently protected neurons against glutamate-induced excitotoxicity. Spinal cord injury is a common condition that is a burden on society and recovery of nerve function is the major objective of therapy. To our knowledge, this is the first study focusing on the interaction between Drp1 and Bax and their colocalization in mitochondria. This colocalization is a potential target for the treatment of spinal cord injury.
Author contributions: Professor WHC and JF conceived and designed the study, and were responsible for fundraising, data acquisition, paper development and writing. KZ, HYY, PYT, WL, YJL, BL, JY, TJ, and JC participated in data acquisition, paper development, writing and authorization. All authors approved the final version of this paper.
Conflicts of interest: None declared.
Financial support: This study was supported by the National Natural Science Foundation of China, No. 81371967 and 81401807; a grant from the 5thPhase of “Project 333” of Jiangsu Province of China, No.BRA2016512; and a grant from the Six Talent Peaks Project of Jiangsu Province of China, No. 2014-WSN-012. The funding bodies played no role in the study design, in the collection, analysis and interpretation of data, in the writing of the paper, and in the decision to submit the paper for publication.
Institutional review board statement: This study was approved by Institutional Animal Care and Use Committee of Nanjing Medical University of China (approval No. 1601036).
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open access statement: This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike 4.0 License, which allows others to remix, tweak,and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- Seeing the wood for the trees: towards improved quantification of glial cells in central nervous system tissue
- 3′-Daidzein sulfonate sodium protects against memory impairment and hippocampal damage caused by chronic cerebral hypoperfusion
- The novel chalcone analog L2H17 protects retinal ganglion cells from oxidative stress-induced apoptosis
- Treatment with NADPH oxidase inhibitor apocynin alleviates diabetic neuropathic pain in rats
- Saikosaponin a increases interleukin-10 expression and inhibits scar formation after sciatic nerve injury
- Various changes in cryopreserved acellular nerve allografts at −80° C