
Schwann cell Myc-interacting zinc-finger protein 1 without pox virus and zinc finger: epigenetic implications in a peripheral neuropathy
2018-08-09 01:27:24DavidFuhrmannHansPeterElssser
中国神经再生研究(英文版) 2018年9期
David Fuhrmann, Hans-Peter Elsässer
Department of Cytobiology and Cytopathobiology, Philipps University of Marburg, Marburg, Germany
Abstract Functionality of adult peripheral nerves essentially relies on differentiation of Schwann cells during postnatal development, as well as fine-tuned re- and transdifferentiation in response to peripheral nerve injury. Epigenetic histone modifications play a major role during the differentiation of embryonic stem cells and diverse organ specific progenitor cells, yet only little is known about the epigenetic regulation of Schwann cells. Just recently,Fuhrmann et al. reported how the transcription factor Myc-interacting zinc-finger protein 1 (Miz1) might contribute to Schwann cell differentiation through repression of the histone demethylase Kdm8. Here, we discuss the potential novel role of Miz1 in Schwann cell differentiation and give a short overview about previously reported histone modifications underlying peripheral nerve development and response to injury.
Key Words: Schwann cell; differentiation; peripheral nerve injury; histone methylation; epigenetics; Miz1
The deletion of Myc interacting zinc finger protein 1 (Miz1)causes a late onset neuropathy with a spontaneous remission(Fuhrmann and Elsässer, 2015; Sanz-Moreno et al., 2015).Miz1 is a zinc finger transcription factor of 794 amino acids(Peukert et al., 1997) which preferentially binds to a consensus sequence near the initiation region of transcription (Wolf).Binding of other proteins to Miz1 can either stimulate (e.g.,Npm, p300, Smad3/4) or inhibit (e.g., Myc, Gfi1, TopBP,Bcl 6) its transactivating function (Herkert and Eilers, 2010).About the first 100 N-terminal amino acids comprise a poxvirus and zinc finger (POZ) domain (Bardwell and Treisman,1994), while 13 zinc finger (ZF) motifs are scattered over the mid and C-terminal part of the protein with a larger gap between ZF12 and ZF13.
Miz1 binds to DNA as a homotetramer and tetramerisation depends on the POZ domain (Stead and Wright, 2014). Consequently, the deletion of the Miz1 POZ domain abrogates its function as a transcription factor. In a Cre/lox mouse model(Gebhardt et al., 2007) the Miz1ΔPOZ genotype was introduced into Schwann cells expressing the Cre recombinase under the control of the desert hedgehog (dhh) promotor(Sanz-Moreno et al., 2015). Mice with Miz1ΔPOZ Schwann cells developed a late onset demyelinating neuropathy with severe hind leg paresis around day 90 after birth (P90), with a spontaneous remission of the clinical symptoms from P120 on(Fuhrmann and Elsässer, 2015; Sanz-Moreno et al., 2015).
Tracing back the development of the Miz1ΔPOZ neuropathy, Fuhrmann et al. (2018) have shown in a recent paper that the upregulation of demyelinating genes and the repression of myelin genes started not earlier than at P60, and that morphological hallmarks like myelin loops, tomacula, degradation of myelin by Schwann cell autophagy or macrophages were not visible ultrastructurally before P60. This means that between the completion of the postnatal development of the peripheral nervous system (PNS) at P30 and the occurrence of the Miz1ΔPOZ phenotype at P60 no obvious pathological features have been observed. As expected from these findings, an RNAseq analysis at P30 revealed a small group of deregulated genes, most likely involved in the very initial pathogenesis of the Miz1ΔPOZ neuropathy. Strikingly, using gene ontology analysis, a strong correlation of these genes with the regulation of the cell cycle and cell proliferation became evident (Fuhrmann et al., 2018).
Usually the proliferation of Schwann cell precursors and Schwann cells peaks around birth, ceases during the postnatal PNS development and is completely shut off around P30(Brown and Asbury, 1981; Stewart et al., 1993). In contrast, the amount of cycling Miz1ΔPOZ Schwann cells was significantly elevated at P30, compared with control Schwann cells, and the percentage of cycling Miz1ΔPOZ Schwann cells continuously rose until P90 and reached up to 3–4% of the total Schwann cell population (Fuhrmann et al., 2018). This confirmed that the upregulation of the cell cycle relevant genes was associated with a progressive increase in Schwann cell proliferation. So far, it is not clear whether there is a subpopulation of Schwann cells which remains in the cell cycle and expands, or whether already arrested Schwann cells reenter the cell cycle. Interestingly, although Schwann cell proliferation was still present in older Miz1ΔPOZ animals, it was 4–5 fold lower than at P90,indicating that the spontaneous remission and regeneration is accompanied by a reduction in the proliferation phenotype.
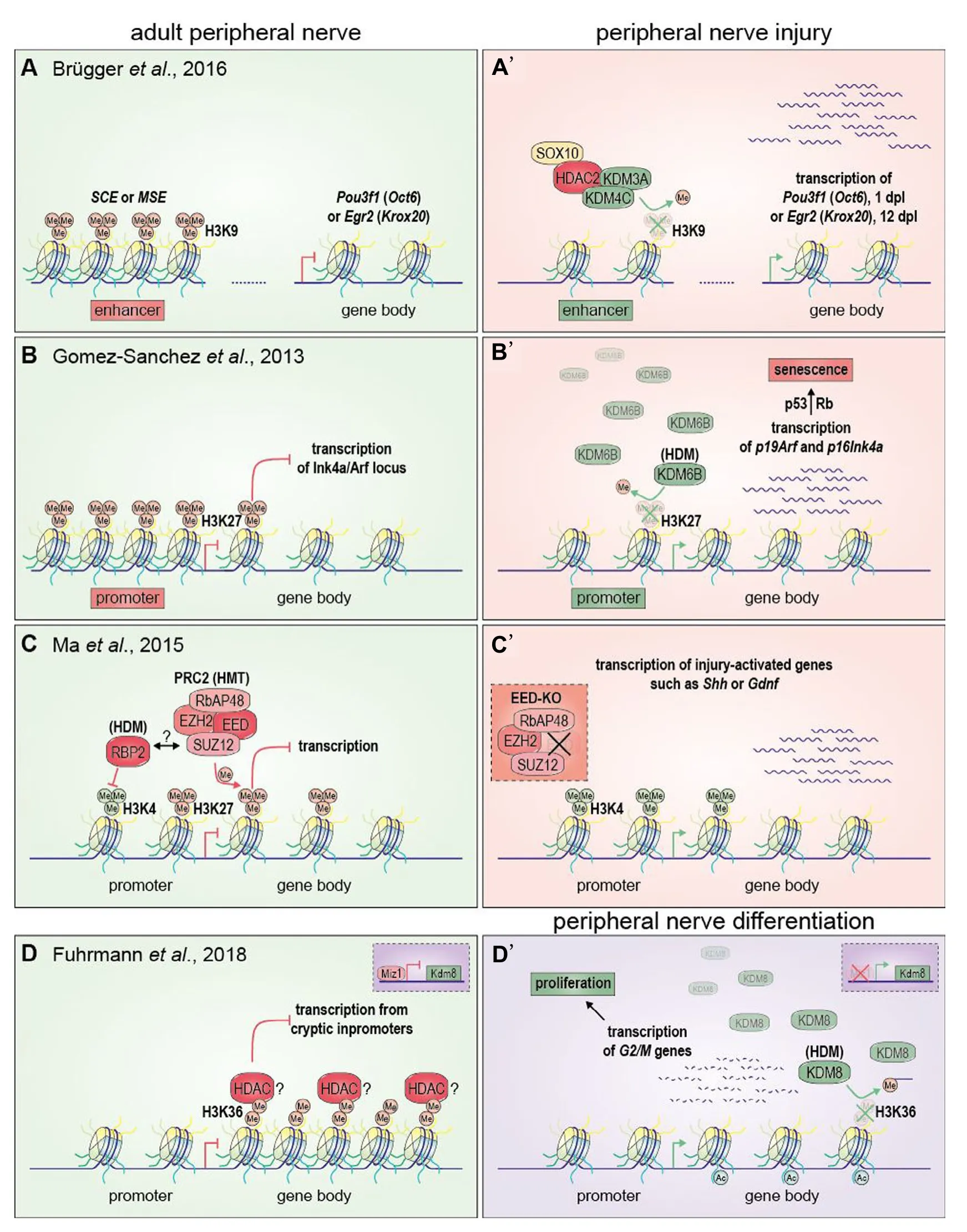
Figure 1 Implications of histone methylation in Schwann cells upon peripheral nerve injury and development.
When testing which of the genes deregulated at P30 in Miz1ΔPOZ animals are direct target genes of Miz1, chromatin immunoprecipitation (ChIP) analysis exposed the two genes Kdm8 and Znhit3 as directly bound by Miz1 at the transcription start site, with Znhit3 as the only gene which was downregulated (Fuhrmann et al., 2018). Znhit3, the mammalian homolog of the yeast Hit1p, is part of the assembly pathway of Box C/D small nucleolar ribonucleoparticles (snoRNPs)(Bizarro et al., 2014) which are involved in post-transcriptional rRNA modifications like 2′-O-methylation of ribose (Watkins and Bohnsack, 2012), but its function in Schwann cells remains to be elucidated. Lysin (K-) demethylase 8 or Jumonji C domain-containing demethylase 5 (Kdm8/Jdmd5) is a histone H3 demethylase with specificity for H3K36me2 (Klose et al.,2006; Kooistra and Helin, 2012). This demethylase has been described in the context of G2/M cell cycle transition, promoting the proliferation of cells by enhancing the expression of Cyclin A1 (Hsia et al., 2010), by repression of Cdkn1b (encoding p21cip) (Ishimura et al., 2012) or by negatively regulating p53 function (Huang et al., 2015). In control Schwann cells Kdm8 expression was continuously downregulated during the postnatal development, parallel to the shutdown of proliferation (Fuhrmann et al., 2018). In contrast, in Miz1ΔPOZ Schwann cells expression of Kdm8 progressively increased until P30 (Fuhrmann et al., 2018). These data, together with the observation that cells from the Schwann cell line mSC80 proliferated faster in the presence of Kdm8, allow the hypothesis, that under normal conditions Kdm8, and hence H3K36 demethylation, is involved in the exit of Schwann cells from the cell cycle. Postnatal repression of Kdm8 was accompanied by a peak expression of Zbtb17 (encoding Miz1) suggesting that Miz1 participates in Kdm8 repression. Taken together, the scenario of the Miz1ΔPOZ neuropathy could start with an insufficient repression of Kdm8 during the postnatal development of the PNS, leading to an impaired cell cycle exit in a subset of Schwann cells which consequently keep on proliferating after P30. Alternatively, reduced H3K36me2 could favor a G0/G1transition in already arrested Miz1ΔPOZ Schwann cells.
The exact link between consistently cycling Schwann cells and the onset of demyelination is not clear. However, during Wallerian regeneration after nerve injury, repair Schwann cells,which exhibit a similar phenotype as Miz1ΔPOZ Schwann cells (Fuhrmann and Elsässer, 2015; Sanz-Moreno et al.,2015), induce dedifferentiation and demyelination of Schwann cells which are located distally from the initial injury site, indicating that repair Schwann cells can induce dedifferentiation in a paracrine mode (Jessen et al., 2015). Following this notion,the onset of the Miz1ΔPOZ neuropathy might occur when a certain threshold of cycling Schwann cells is achieved, explaining the late onset of the Miz1ΔPOZ neuropathy. In this model Miz1 is necessary to repress Kdm8 expression during the postnatal development. As mentioned above, Miz1 represses genes in cooperation with different binding partners, but the corepressor in Schwann cells is still unknown.
A variety of epigenetic modifications have been shown in Schwann cells during development and nerve injury, concerning histone acetylation/deacetylation, histone methylation/demethy-lation and DNA methylation/demethylation (Jacob, 2017; Ma and Svaren, 2018). Specifically, in response to nerve injuries, the Jmjc domain histone demethylases Kdm3A and Kdm4c build a complex with the histone deacetylase 2(Hdac2) and the transcription factor Sox10. This protein complex demethylates the repressive H3K9me2/3 histone mark at an enhancer element of Pou3f1 (Oct6) one day post lesion (dpl), leading to a higher expression of Oct6 and, subsequently, to an inhibition of Jun expression and attenuated demyelination (Brügger et al., 2017) (Figure 1A, A’). On the other hand, the same complex also de-represses the Egr2(Krox20) enhancer at 12 dpl and promotes P0 re-expression and remyelination. Strikingly, this indicates that recruitment of histone deacetylases, such as Hdac2, does not always correlate with transcriptional repression. Furthermore, the proliferation of Schwann cells is limited upon oncogenic stimuli or nerve injury by the demethylation of H3K27me2/3 at the promotors of p19Arf,p16Ink4aand p15Ink4bAby enhancing the expression of these cell cycle inhibitor genes and by induction of senescence (Gomez-Sanchez et al., 2013) (Figure 1B, B’). Similar to histone demethylases (HDM) which function as erasers on histone methyl modifications, histone methyl transferases(HMT) can play a part as writers in regulating chromatin. For example the polycomb repression complex 2, which catalyzes the methylation of H3K27, is part of an epigenetic pathway that represses genes which are not expressed in Schwann cells(such as Shh) or only during Schwann cell differentiation (such as Gdnf) (Ma et al., 2015) (Figure 1C, C’). Prc2 establishes the repressive H3K27me3 mark in promoter regions and the gene body. Simultaneously, the promoter regions also exhibit active H3K4me3 marks, which are possibly restrained to promoter regions by the HDM Rbp2, which is able to interact with Prc2 (Pasini et al., 2008). Following nerve injury or knock out of an essential Prc2 subunit, the H3K27me3 repressive mark is lost resulting in increased H3K4 trimethylation and expression of repair Schwann cell specific genes (e.g., Shh or Gdnf).The function of Kdm8 for the proliferation of Schwann cells during development by controlling the H3K36 methylation status adds a new facet of epigenetic regulation in this cell type(Fuhrmann et al., 2018) (Figure 1D, D’). The work indicates,that Kdm8 demethylates H3K36me2, which is increasingly distributed in the distal 3’ gene body of cell cycle associated genes (e.g., Psrc1, Iqgap3, Top2a or Ckap2), similar to the distribution observed for H3K36me3 (Carrozza et al., 2005;Keogh et al., 2005). According to the established mode of transcriptional regulation (studied in yeast) H3K36 is trimethylated by Setd2, which interacts with the RNA-Polymerase, during the process of active transcription. Subsequently, H3K36me3 recruits HDACs thereby introducing a closed chromatin state to prevent spurious transcription from cryptic promoters.H3K36me2 might have a preceding function because trimethylation of H3K36 depends on the presence of H3K36me2. Demethylation of H3K36me2 may prevent HDAC recruitment and favor transcription of cell cycle associated genes during development.
The function of H3K36 methylation in Schwann cell biology and the pathogenesis of neuropathies is unknown and the impact of Kdm8 overexpression in Schwann cells for the development of a demyelinating neuropathy needs further investigation using appropriate mouse models. Also, so far Kdm8 mutations or variations have not been described in Charcot-Marie-Tooth (CMT) patients. However, as men-tioned above, other histone 3 methylations have been shown to be involved in the regulation of proliferation and differentiation of Schwann cells (Gomez-Sanchez et al., 2013; Ma et al.,2015; Brügger et al., 2017). Moreover, in cancer cells, derived from other cell types, cancer cell behavior has been linked to epigenetic modifications of histone H3 including its methylation status. Histone demethylases are now targets for therapeutic approaches, e.g., in acute myeloid leukemia (Castelli et al., 2018), and a variety of inhibitors have been developed for the monoamino oxidase like lysine-specific demethylase 1 and 2 (LSD1 and 2/Kdm1A and B) and the Jumonji C (JmjC)histone demethylases (Jambhekar et al., 2017). A few of them have already been used in mouse models and the best documented inhibitor is probably GSK-J1/4 developed with its highest specificity for KDM5B, KDM5C, KDM6A, KDM6B(Kruidenier et al., 2012). GSK-J1/4 has successfully been used for the reduction of human glioma tissue in a mouse xenograft model (Hashizume et al., 2014) demonstrating the potential of Kdm inhibitors for therapeutic in vivo approaches, suggesting that Kdm8 could become a new target in the therapy of neuropathies.
Author contributions: DF and HPE wrote the text. DF made the figure.Conflicts of interest: None declared.
Financial support: This work was supported by Deutsche Forschungsgemeinschaft (DFG grant EL125/6-1).
Copyright transfer agreement: The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open access statement: This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike 4.0 License, which allows others to remix, tweak,and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- Seeing the wood for the trees: towards improved quantification of glial cells in central nervous system tissue
- 3′-Daidzein sulfonate sodium protects against memory impairment and hippocampal damage caused by chronic cerebral hypoperfusion
- The novel chalcone analog L2H17 protects retinal ganglion cells from oxidative stress-induced apoptosis
- Treatment with NADPH oxidase inhibitor apocynin alleviates diabetic neuropathic pain in rats
- Saikosaponin a increases interleukin-10 expression and inhibits scar formation after sciatic nerve injury
- Various changes in cryopreserved acellular nerve allografts at −80° C