
Role of nuclear factor κB in multiple sclerosis and experimental autoimmune encephalomyelitis
2018-08-09 01:27YuanYueSarrabethStoneWenshengLin
中国神经再生研究(英文版) 2018年9期
Yuan Yue , Sarrabeth Stone , Wensheng Lin ,
1 Department of Neuroscience, University of Minnesota, Minneapolis, MN, USA
2 Institute for Translational Neuroscience, University of Minnesota, Minneapolis, MN, USA
Abstract The transcription factor nuclear factor κB (NF-κB) plays major roles in inflammatory diseases through regulation of inflammation and cell viability. Multiple sclerosis (MS) is a chronic inflammatory demyelinating and neurodegenerative disease of the central nervous system (CNS). It has been shown that NF-κB is activated in multiple cell types in the CNS of MS patients, including T cells, microglia/macrophages, astrocytes, oligodendrocytes, and neurons. Interestingly, data from animal model studies, particularly studies of experimental autoimmune encephalomyelitis, have suggested that NF-κB activation in these individual cell types has distinct effects on the development of MS. In this review, we will cover the current literature on NF-κB and the evidence for its role in the development of MS and its animal model experimental autoimmune encephalomyelitis.
Key Words: multiple sclerosis; experimental autoimmune encephalomyelitis; nuclear-factor κB; T cell;macrophage; microglia; astrocyte; oligodendrocyte; neuron
Introduction
Multiple sclerosis (MS) is a chronic inflammatory, demyelinating, and neurodegenerative disease characterized by central nervous system (CNS) inflammation, demyelination,oligodendrocyte death, axon degeneration, and neuron loss(Noseworthy et al., 2000; Frohman et al., 2006; Ontaneda et al., 2017). MS is believed to be caused by a T cell-mediated autoimmune reaction against oligodendrocytes and myelin. MS is a complex disease involving the interplay of inflammatory cells (T cells, B cells, and macrophages) and CNS resident cells(microglia, astrocytes, oligodendrocytes, and neurons). It is generally believed that, during MS pathogenesis, inflammation causes tissue damage, including oligodendrocyte death, demyelination, axon degeneration, and neuron death. Subsequently,the tissue damage promotes inflammation, forming a vicious cycle. However, besides this “outside-in” theory (in which T cells become activated in the periphery and then invade the CNS), some other studies have argued that the initiation of disease pathology occurs within the CNS. This “inside-out”hypothesis suggests that MS is initiated by primary degeneration of oligodendrocytes and myelin, resulting in the release of antigenic myelin components, which elicits an autoimmune reaction against myelin components (Stys et al., 2012; Traka et al., 2016). Experimental autoimmune encephalomyelitis (EAE)is the most commonly used animal model in MS studies, and shares many clinical, pathological, and immunological characteristics with MS, such as inflammation, demyelination, oligodendrocyte loss, axon loss, and neuron loss (Constantinescu et al., 2011; Stanojlovic et al., 2016; Lassmann and Bradl, 2017).Nuclear factor κB (NF-κB) is one of the master transcription factors that regulate the activity of inflammatory cells such as T cells, macrophages, and microglia. Recent studies have also shown that NF-κB regulates the viability of resident cells in inflammatory lesions (Mc Guire et al., 2013; Park and Hong,2016). Importantly, it has been shown that NF-κB is activated in many cell types in MS and EAE, including T cells, microglia/macrophages, astrocytes, oligodendrocytes, and neurons(Gveric et al., 1998; Bonetti et al., 1999; Yan and Greer, 2008;Mc Guire et al., 2013). In this review, we discuss the current understanding of the contribution of NF-κB activation in these individual cell types to the development of MS and EAE.
NF-κB Signaling
NF-κB is a hetero or homodimer of the Rel family of proteins, which includes RelA (p65), RelB, c-Rel, NF-κB1 (p105),and NF-κB2 (p100) (Hayden and Ghosh, 2012). The p105 and p100 proteins are processed into their shorter forms, p50 and p52, respectively, by the post-translational cleavage of the C-terminal region containing ankyrin repeats. This family of proteins has the following in common: an N-terminal 300 amino acid Rel homology domain, which is responsible for sequence-specific DNA binding, dimerization, and inhibitory protein binding (Huxford and Ghosh, 2009). These proteins also contain the nuclear localization sequence (NLS), which is responsible for NF-κB nuclear translocation (Oeckinghaus and Ghosh, 2009). The structure of the transcription activation domain (TAD) is responsible for transcriptional activity after NF-κB activation. Depending on the TAD structure, NF-kB family members can be classified into subfamily I and subfamily II. Subfamily I includes the proteins RelA, c-Rel, and RelB,which contain a TAD. In contrast, subfamily II includes the proteins p52 and p50, which do not contain the TAD and are therefore unable to activate gene transcription following NF-κB activation (Mincheva-Tasheva and Soler, 2013). The NF-κB family proteins can assemble into several homodimeric and heterodimeric species, of which the most abundant dimer formed in mammals is the p65/p50 heterodimer. In addition,other homo- or heterodimers, including the p50/p50 and p52/p52 homodimers, and the RelA/p52, RelB/p50, and RelB/p52 heterodimers, exist and have distinct functions (Smale, 2012;Christian et al., 2016; Collins et al., 2016).
Under resting conditions, the NF-κB dimers are mainly sequestered in the cytoplasm by binding to the “inhibitor of κB” (IκB) family (IκBα, IκBβ, IκBε, IκBγ,IκBδ, and Bcl-3), of which IκBα is the best characterized(Hayden and Ghosh, 2008). Through their ankyrin repeat domains IκB family proteins bind to NF-κB proteins, masking their NLS and keeping them sequestered and inactive in the cytoplasm. Because of the ankyrin repeat domains in their C-terminal halves, NF-κB1 (p105) and NF-κB2 (p100) also function as IκB proteins (Park and Hong, 2016). NF-κB becomes activated following IκBα phosphorylation at the Ser 32 and Ser 36 residues of the ankyrin repeats, leading to the release of IκBα from the NF-κB complex (Perkins, 2012).However, IκBα is one of the first genes transcribed following NF-κB activation; thus, the reduction of IκBα protein levels in the cytoplasmic is temporary and increases rapidly after activation in response to the newly synthesized IκBα (Place et al., 2001; Mincheva-Tasheva and Soler, 2013). Extracellular stimuli, such as proinflammatory cytokines, bacteria, viruses, physiological stress, oxidative stress, modified proteins,apoptotic mediators or chemical agents can trigger NF-κB activation (Yan and Greer, 2008; Shih et al., 2015). In general,NF-κB can be activated through three distinct pathways: the canonical/classical pathway, non-canonical/alternative pathway, and atypical pathway (Figure 1).
The canonical NF-κB pathway, also called the classical pathway, can be induced via the activation of a variety of cell surface receptors, such as the interleukin (IL)-1 receptor (IL-1R), Toll-like receptors (TLRs), TNF receptor, or T and B cell receptors (TCR and BCR). Stimulation through these receptors binding with their ligands leads to the activation of the IκB kinase (IKK) complex, a trimeric complex composed of two catalytic subunits, IKKα/IKK1 and IKKβ/IKK2, and a NF-κB essential modulator regulatory subunit (NEMO/IKKγ) (Vallabhapurapu and Karin, 2009). IKK then phosphorylates the IκB proteins at the Ser 32 and Ser 36 residues of the ankyrin repeats, leading to rapid polyubiquitination and degradation by the proteasome, and subsequent release of NF-κB (Hayden and Ghosh, 2008; Schmid and Birbach, 2008).Following the degradation of IκB, NF-κB translocates to the nucleus, where it binds to DNA and activates downstream gene transcription (Oeckinghaus et al., 2011). The heterodimer of p65 and p50 is the form of NF-κB most commonly activated by the canonical pathway (Hayden and Ghosh, 2011).However, NF-κB activation also leads to newly synthesized IκBα, which then re-inhibits NF-κB and forms an auto feedback loop (Park and Hong, 2016). In the canonical pathway, IKKβ is required and plays a key role in the rapid degradation of NF-κB-bound IκB dimers, which occurs within minutes. IKKα is redundant in this pathway and controls the processing of p100, which requires several hours (Maruyama et al., 2010; Ben-Neriah and Karin, 2011). However, IKKα is also capable of activating the canonical NF-κB pathway when IKKβ is limited (Lam et al., 2008). Moreover, the canonical NF-κB pathway is indispensable for immune responses, both acute and chronic. Additionally, the canonical pathway plays vital roles in many important biological processes, including activation of immune cells (such as dendritic cells), survival and maturation of B-cells, lymphoid organogenesis, and bone metabolism (Ben-Neriah and Karin, 2011; Park and Hong,2016).
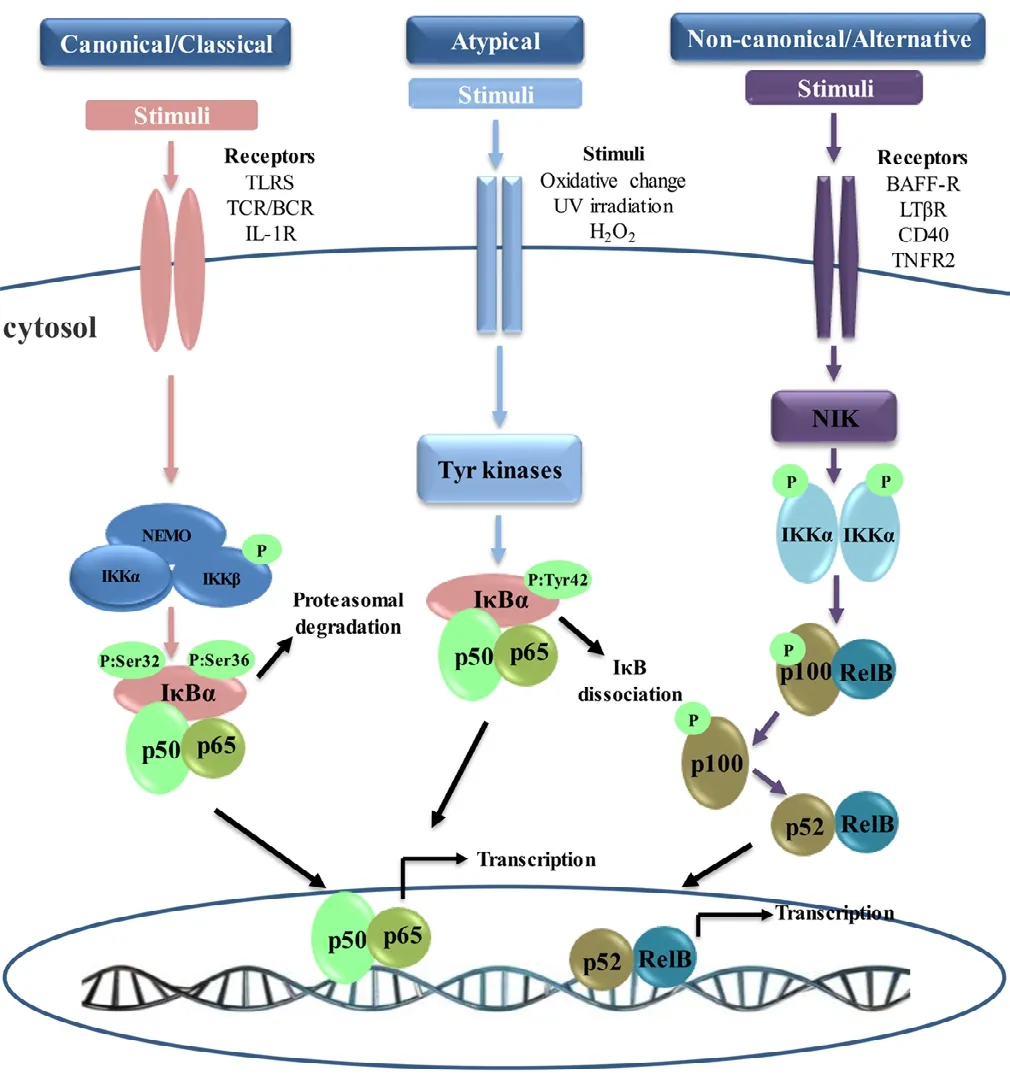
Figure 1 Nuclear factor κB (NF-κB) activation signaling pathways.Canonical NF-κB pathway: the canonical pathway is inhibitor of κB(IκB) kinase (IKK)-dependent and IκB-dependent. Stimulation through receptors such as Toll-like receptors (TLRs) and T cell receptors (TCRs)leads to the activation of IKK complex, which phosphorylates IκBα and leads to its proteasomal degradation. Following the degradation of IκBα,heterodimer p65/p50 translocates to the nucleus and activates downstream gene transcription. Non-canonical NF-κB pathway: the non-canonical pathway is strictly dependent on IKKα, but independent of IKKβ, NF-κB essential modulator regulatory subunit (NEMO), and IκBα. Ligand-induced activation of receptors such as CD40 leads to the activation of NF-κB-inducing kinase (NIK). NIK specifically phosphorylates and activates IKKα homodimers. IKKα phosphorylation induces the polyubiquitination and proteasomal degradation of the NF-κB2 (p100) precursor to its active form, p52. Then, the newly formed heterodimer RelB/p52 translocates to the nucleus, resulting in the transcription of target genes.Atypical NF-κB Pathway: the atypical pathway is IKK-independent but IκBα dependent. This pathway is usually activated by the stimulation of the tyrosine kinase receptor or after oxidative challenge. After stimulation,IκBα is phosphorylated on Tyr 42 or on serine residues in the IκBα PEST domain. IκBα phosphorylation leads to the release of p65/p50,which translocates to the nucleus and transcribes the target genes. BCR:B cell receptors; IL-1R: interleukin-1 receptor; UV: ultraviolet; BAFF-R:B-cell activating factor receptor belonging to tumor necrosis factor family receptor; LTβR: lymphotoxin β-receptor; TNFR2: tumor necrosis factor receptor 2.
The non-canonical NF-κB pathway, also called the alternative pathway, can be activated by a subset of TNF-receptor superfamily members, such as B-cell activating factor receptor belonging to tumor necrosis factor (TNF) family receptor(BAFF-R), lymphotoxin β-receptor (LTβR), CD40, receptor activator for nuclear factor κB (RANK), TNF receptor 2 (TNFR2), and Fn14 (Sun, 2011; 2017; Zhang et al., 2017).In addition to the non-canonical NF-κB pathway, these receptors also simultaneously trigger the canonical pathway. In contrast to the canonical NF-κB pathway, the non-canonical pathway is dependent on IKKα, but independent of IKKβ and NEMO (Shih et al., 2015). Ligand-induced activation of these receptors leads to the activation of NF-κB-inducing kinase (NIK). Under normal conditions, NIK is bound by TNF receptor-associated factor-3 (TRAF3), which mediates recruitment of NIK to TRAF2 and results in NIK ubiquitination and continuous degradation (Sun, 2017). Because of the continuous degradation, the endogenous levels of NIK are very low and prevents NIK accumulation or activation of the non-canonical NF-κB pathway (Sun, 2012). However,ligand-induced activation leads to the proteolysis and degradation of TRAF3, resulting in NIK release and accumulation.NIK then specifically phosphorylates and activates IKKα homodimers. IKKα phosphorylation induces the polyubiquitination and proteasomal degradation of the NF-κB2 (p100)precursor to the active form p52 (Maruyama et al., 2010). The newly formed RelB/p52 heterodimers are then translocated to the nucleus, resulting in the transcription of target genes (Vallabhapurapu and Karin, 2009; Sun, 2017). The non-canonical NF-κB pathway plays an important role in several biological functions, including the development of secondary lymphoid organs, and the proliferation, survival, and maturation of B and T cells (Dejardin, 2006). The activation of the non-canonical pathway requires NIK synthesis and accumulation, whereas activation of the canonical pathway is independent of protein synthesis. Thus, activation of the non-canonical pathway is much slower than the activation of the canonical NF-κB pathway (Yan and Greer, 2008; Cildir et al., 2016; Sun, 2017).
In addition to the major canonical and non-canonical pathways, another IKK-independent pathway, the atypical NF-κB signaling pathway, has also been described (Perkins, 2007;Hoesel and Schmid, 2013). The atypical pathway is IKK-independent but IκBα dependent, leading to the translocation of the heterodimer p65/p50 to the nucleus (Mincheva-Tasheva and Soler, 2013). This pathway is usually activated by the stimulation of the tyrosine kinase receptor or following oxidative challenge, pervanadate stimulation, or ultraviolet irradiation(Gloire and Piette, 2009). In response to these types of stimulation, IκBα is phosphorylated on Tyr 42 or on serine residues in the IκBα PEST domain (Gloire and Piette, 2009; Siomek,2012). IκBα phosphorylation leads to the release of p65/p50,which translocates to the nucleus and transcribes the target genes. Comparing the three NF-κB signaling pathways, the canonical and non-canonical pathways are IKK-dependent,but the atypical pathway is IKK-independent. The canonical and atypical pathways are IκB-dependent, while the non-canonical pathway is IκB-independent. The canonical and atypical pathways induce nuclear translocation of heterodimer p65/p50, while the non-canonical pathway induces nuclear translocation of dimer RelB/p52 (Mincheva-Tasheva and Soler, 2013).
The Role of NF-κB Activation in T Cells in MS and EAE
T cells are responsible for a variety of immune responses, including attacking foreign substances, augmentation of the B cell response and production of cytokines that direct responses and activities in other immune cells (Murphy and Weaver,2016). MS and EAE are T cell-mediated autoimmune diseases in the CNS. It is generally thought that MS and EAE are initiated by T cell priming in the periphery immune system. These myelin reactive T cells then migrate across the blood-brain barrier (BBB) and into the CNS where they cause inflammation, and ultimately, demyelination and neurodegeneration(Baxter, 2007; Dendrou et al., 2015; Prinz and Priller, 2017).NF-κB plays a crucial role in various aspects of T cell development and function, and it predominantly mediates through RelA/p50 and p50/c-Rel heterodimers or p50/p50 homodimers by stimulating the T cell receptor or co-receptor CD28(Schmidt-Supprian et al., 2003; Zheng et al., 2003).
RelA-deficient mice are embryonically lethal due to massive apoptosis that occurs in the liver (Beg et al., 1995). RelA-deficient T cells develop normally into mature T cells and express similar levels of IL-2 in comparison with control cells after stimulation by various agents. However, T cells lacking RelA show a significant reduction in proliferative responses following stimulation by various agents (Doi et al., 1997).RelA-deficient T cells are significantly compromised in Th17 differentiation and have decreased numbers of γδ T cells expressing IL-17 (Powolny-Budnicka et al., 2011; Ruan et al.,2011). Moreover, RelA deficiency in T cells significantly reduces the number of NKT cells in the bone marrow, spleen,liver and thymus by preventing the NK1.1–to NK1.1+transition and impairing the proliferation of NKT-cell precursors driven by IL-7 and IL-15. (Vallabhapurapu et al., 2008). In addition, RelA is also a critical regulator of the survival of proliferating CD8 T cells and the development of effector Treg cells (Mondor et al., 2005; Vasanthakumar et al., 2017). Mice lacking the p50 subunit of NF-κB develop normally to acquire mature T cells and a structurally intact immune system.However, these mice show defects in their immune response,such as decreased serum immunoglobulin and defective responses to different pathogens (Sha et al., 1995). Furthermore,NF-κB1-deficient mice are resistant to EAE in that they have a reduced incidence and disease severity, and attenuated CNS inflammation (Hilliard et al., 1999). T cells from mice lacking NF-κB1 were found to have impaired differentiation of myelin oligodendrocyte glycoprotein (MOG)-specific T cells to Th1 or Th2 effector cells, thus rendering the mice resistant to EAE (Hilliard et al., 1999). Finally, mice lacking the c-Rel subunit of NF-κB develop normally to acquire mature T cells and a structurally intact immune system. However, T cells from c-Rel-knockout mice exhibit proliferative defects after stimulation by various agents and fail to differentiate into functional effector cells CD8+or CD4+T Cells, which is associated with impaired cytokine production, especially that of IL-2(Kontgen et al., 1995; Liou et al., 1999). c-Rel-knockout mice have significantly reduced numbers of thymic and peripheral Treg cells due to a hemopoietic defect, suggesting the importance of c-Rel in the development of Treg cells in the thymus and the maintenance peripheral Treg cells (Chen et al., 2011).c-Rel-deficient T cells also have deficiencies in Th17 differentiation, which is similar to the phenotype of RelA-deficient T cells (Chen et al., 2011). In addition, c-Rel-deficient mice have significant defects in Th1 responses, which are attributed to the selective blockade of IL-12 production and the abrogation of interferon-γ (IFN-γ) expression (Hilliard et al., 2002). In agreement with these results, mice lacking c-Rel are resistant to EAE in that they have a reduced incidence of EAE, clinical score, and degree of CNS inflammation (Hilliard et al., 2002;Chen et al., 2011).
In addition to the NF-κB family subunits, the IKK complex has also been suggested to be essential in driving inflammatory immune responses in MS and EAE. Studies assessing the T cell-specific deletion of IKKβ have shown that IKKβ is dispensable for the survival of naive peripheral T cells but cause a severe deficiency in regulatory and memory T cell populations.In contrast, other studies have shown that deletion of NEMO or replacement of IKKβ with a kinase-dead mutant prevents peripheral T cells development altogether, which is not compatible with the generation and/or persistence of mature T cell(Schmidt-Supprian et al., 2003). Moreover, IKKβ-deficient T cells can produce similar cytokines as control cells and differentiate into effector T cells, which suggests that IKKβ is dispensable for T cell activation. However, T cells lacking IKKβ are defective in recall responses after immunizations under Th1- or Th2-polarizing conditions (Schmidt-Supprian et al., 2004). Importantly, deficiency of IKKβ specifically in T cells significantly impairs MOG35-55-specific responses in T cells and completely protects against the induction of EAE in C57BL/6 mice. This results in a reduced incidence of EAE,clinical score, and degree of CNS inflammation (Greve et al.,2007). In conclusion, NF-κB plays a crucial role in the development and differentiation of T cells, and NF-κB activation in T cells promotes the development of MS and EAE.
The Role of NF-κB Activation in Microglia/Macrophages in MS and EAE
Microglia and macrophages are the primary sensors of CNS pathology and are recruited rapidly to sites where trauma, infection, or autoimmune inflammation is taking place. During MS and EAE pathogenesis, microglia/macrophages are activated by Th1 cytokines produced by myelin reactive T cells. Activated microglia/macrophages produce large quantities of inflammatory mediators, including pro-inflammatory cytokines,chemokines, reactive oxygen species (ROS), and reactive nitrogen species (RNS), which amplify inflammation and lead to demyelination and neurodegeneration in the CNS (Chitnis and Weiner, 2017; O’Loughlin et al., 2018). NF-κB is activated in microglia/macrophages in MS and EAE, and NF-κB activation in microglia/macrophages enhances the production of inflammatory mediators that further activate microglia/macrophages, forming a vicious cycle (Blank and Prinz, 2014; Leibowitz and Yan, 2016). Mice with IκBα knockout specifically in myeloid cells, which are characterized by the constitutive activation of NF-κB in microglia/macrophages during EAE,develop an exacerbated EAE disease course with enhanced inflammatory infiltration and demyelination in the CNS (Ellrichmann et al., 2012). Furthermore, IKKβ deficiency specifically in myeloid cells attenuates disease severity as well as demyelination in the CNS, and reduces leukocyte infiltration, inflammatory gene expression, and encephalitogenic T cell activation in the CNS of mice undergoing EAE. In addition, mice with IKKβ deficiency specifically in myeloid cells also reduce BBB permeability in the spinal cord while undergoing EAE (Hao et al., 2016; Lee et al., 2016). These data suggest that the IKKNF-κB pathway-induced myeloid cell activation contributes to EAE pathogenesis. Moreover, conditional depletion of microglial transforming growth factor (TGF)-b-activated kinase 1 (TAK1), which blocks NF-κB activation in microglia,protects the mice against EAE by significantly reducing CNS inflammation and diminishing axonal and myelin damage(Goldmann et al., 2013). The results of these studies suggest that NF-κB activation in microglia/macrophages promotes the development of MS and EAE.
The Role of NF-κB Activation in Astrocytes in MS and EAE
Astrocytes are the most abundant and heterogeneous glial cell in the CNS. Astrocytes become activated and play an important role in MS by contributing to demyelination and axonal degeneration and creating a permissive environment that promotes remyelination (Correale and Farez, 2015; Domingues et al., 2016; Ludwin et al., 2016). Activated astrocytes show increased NF-κB activation during MS and EAE (Brambilla et al., 2009; Blank and Prinz, 2014). Transgenic GFAP/IκBαΔN mice express IκBαΔN, a deletion mutant lacking the N-terminal 36 amino acids of IκBα that functions as a dominant inhibitor of NF-κB signaling, specifically in astrocytes under the control of the glial fibrillary acidic protein(GFAP) transcriptional control region, resulting in NF-κB inactivation in astrocytes (Krappmann et al., 1996; Bracchi-Ricard et al., 2008). These mice develop normally and appear healthy. However, NF-κB inactivation specifically in astrocytes leads to learning and memory deficits and impairs longterm potentiation (LTP) in the hippocampal CA1 region in female mice, but has no effect on male mice (Bracchi-Ricard et al., 2008). Interestingly, NF-κB inactivation specifically in astrocytes attenuates EAE disease severity and improves functional recovery following EAE. GFAP/IκBαΔN mice undergoing EAE exhibit a dramatic reduction in the expression of pro-inflammatory cytokines, chemokines and adhesion molecules in the CNS, but show increased leukocyte infiltration into the CNS (Brambilla et al., 2009). A study using GFAP/IκBαΔN mice also showed that NF-κB inactivation in astrocytes protects mice from optic nerve damage and reduces the loss of retinal ganglion cells in experimental optic neuritis(Brambilla et al., 2012). Moreover, another study showed that NF-κB inactivation specifically in astrocytes attenuates inflammation and demyelination in the cuprizone model (Raasch et al., 2011).
Furthermore, one study showed that laquinimod, an oral immunomodulatory compound, suppresses astrocytic NF-κB activation in vitro and in vivo, and that laquinimod treatment protects mice from cuprizone-induced demyelination (Bruck et al., 2012). On the other hand, Oeckl et al. (2012) generated a transgenic mouse model that expresses a constitutively active IKKβ (IKKβ-CA) in astrocytes under control of the GFAP promotor. Interestingly, IKKβ-CA expression specifically in astrocytes results in NF-κB activation and causes vast neuroinflammation, which is associated with increased levels of inflammatory cytokines, activated microglia, and astrogliosis.Taken together, these studies suggest that activation of astrocytic NF-κB facilitates inflammation and demyelination in MS and EAE.
The Role of NF-κB Activation in Oligodendrocytes in MS and EAE
The primary function of oligodendrocytes is to produce and maintain the myelin sheath that insulates and protects CNS axons. Evidence suggests that NF-κB is not involved in myelin formation and maintenance in the CNS under normal conditions (Blank and Prinz, 2014; Kretz et al., 2014). Mice with a deletion in the gene encoding either c-Rel, RelB, or p52 do not exhibit any myelin defects in the CNS (Hilliard et al., 1999,2002). Moreover, mice with a CNS-restricted deletion of either p65, IKKα, IKKβ, or IKKγ do not show any myelin abnormalities in the CNS (van Loo et al., 2006; Kretz et al., 2014).However, one study has shown that impaired NF-κB activity is associated with myelin abnormalities in the CNS of patients with a duplication of Xq28, a X-linked intellectual disability syndrome characterized by 0.5-Mb duplication (hemizygous in males and heterozygous in females) at chromosome Xq28(Philippe et al., 2013). On the other hand, although several in vitro studies have suggested that NF-κB activation is required for Schwann cell differentiation and myelin formation in the peripheral nervous system (PNS), a recent report shows that Schwann cell-restricted deletion of IKKβ has no effect on myelin formation in the PNS in a mouse model (Nickols et al.,2003; Limpert et al., 2013; Morton et al., 2013). Interestingly,a recent study showed that expression of IκBαΔN, a super-suppressor of NF-κB, in oligodendrocytes and Schwann cells had no effect on developmental myelination in the CNS and PNS of young, developing mice or myelin maintenance in fully myelinated adult mice (Stone et al., 2017). Collectively,these studies suggest that NF-κB is dispensable for the myelinating function of oligodendrocytes and Schwann cells under normal conditions.
MS and EAE are initiated by autoimmune attacks against oligodendrocytes and myelin (Bradl and Lassmann, 2010; Stone and Lin, 2015). NF-κB activation has been observed in oligodendrocytes in MS and EAE (Bonetti et al., 1999; Lin et al.,2013). In MS and EAE, inflammatory mediators such as immune cytokines, ROS, RNS are thought to contribute significantly to oligodendrocyte death (Bradl and Lassmann, 2010;Mahad et al., 2015). Several in vitro reports have suggested that NF-κB activation enhances the viability of oligodendrocyte exposed to inflammatory mediators. One study showed that treating rat oligodendrocyte precursor cells (OPCs) with hydrogen peroxide causes cell apoptosis and NF-κB activation(Vollgraf et al., 1999). Another study showed that treatment with medium conditioned by non-activated microglia induces NF-κB activation in OPCs, reduces OPC apoptosis, and enhances OPC differentiation. Interestingly, enhancing NF-κB activation in OPCs by using anti-sense oligonucleotides targeting IκBα also inhibits OPC apoptosis and enhances OPC differentiation (Nicholas et al., 2001). Moreover, one study showed that enhanced expression of either p50 or p65 in a rat oligodendroglial cell line CG-4 cells prevents TNF-α-induced cell apoptosis, with p50 being more effective than p65 in preventing apoptosis (Hamanoue et al., 2004). Furthermore,one study showed that enforced expression of IκBαΔN in a mouse oligodendroglial cell line Oli-neu cells inhibits NF-κB activation and makes the cells sensitive to cytotoxicity caused by immune cytokines, ROS, and RNS (Lin et al., 2012).Additionally, a recent study showed that enforced expression of cellular FLICE (FADD-like IL-1β-converting enzyme)-inhibitory protein (cFLIP), a major responsive gene of NF-κB,protects cultured primary oligodendrocytes against TNF-α cytotoxicity (Tanner et al., 2015).
Recent in vivo studies have suggested that NF-kB activation in oligodendrocytes has cytoprotective effects in MS and EAE.Stone et al. (2017) generated a mouse model that expresses IκBαΔN specifically in oligodendrocytes. They found that IκBαΔN expression specifically in oligodendrocytes selectively blocks activation of NF-κB in oligodendrocytes and results in exacerbated oligodendrocyte death and myelin loss in the CNS of young, developing mice that ectopically express IFN-γ, a key pro-inflammatory cytokine in MS and EAE,in the CNS. Although IκBαΔN expression specifically in oligodendrocytes does not affect oligodendrocyte apoptosis during the demyelination process or oligodendrocyte regeneration during the remyelination process in the cuprizone model, the authors found that inactivation of NF-κB specifically in oligodendrocytes aggravates IFN-γ-induced remyelinating oligodendrocyte apoptosis and remyelination failure in the cuprizone model. Importantly, they found that mice expressing IκBαΔN specifically in oligodendrocytes develop very severe EAE and have a very high rate of mortality, without altering the peripheral immune response. Moreover, another study shows that knockdown of cFLIP using a cFLIP-specific shRNA-expressing virus makes oligodendrocytes sensitive to inflammation in the CNS of transgenic mice that ectopically express human IL-1β in astrocytes (Tanner et al., 2015). In contrast, a previous study showed that deletion of IKKβ in oligodendrocyte, using IKKβFL; MOG/Cre mice, does not affect the development of EAE (Raasch et al., 2011).
It is known that NF-κB can be activated in oligodendrocytes in MS and EAE through both the IKKβ-dependent and IKKβ-independent pathways (Yan and Greer, 2008; Mc Guire et al., 2013; Lin et al., 2012; 2013). Interestingly, recentstudies have suggested that NF-κB can also be activated by pancreatic endoplasmic reticulum kinase (PERK) signaling, an IKKβ-independent pathway, in oligodendrocytes in MS and EAE (Lin et al., 2012, 2013). A large number of studies have demonstrated the critical role of the PERK pathway in oligodendrocytes in myelin disorders, including MS and EAE (Lin et al., 2005, 2006, 2007, 2008, 2013, 2014; Way et al., 2015).The PERK pathway becomes activated following endoplasmic reticulum stress and phosphorylates eukaryotic translation initiation factor 2α (eIF2α); this results in a reduction in global protein synthesis but an increase in the expression of numerous cytoprotective genes (Lin and Popko, 2009; Stone and Lin, 2015). Phosphorylation of eIF2α induces NF-κB activation by repressing the translation of IκBα (Deng et al., 2004). It has been demonstrated that IFN-γ activates the PERK-eIF2α pathway in oligodendrocytes through the JAKSTAT pathway (Lin et al., 2005; Lin and Lin, 2010). One study showed that treatment with IFN-γ also leads to NF-κB activation in Oli-neu cells (Lin et al., 2012). Importantly, this study showed that forced expression of PERKΔC, a dominant inhibitor of PERK, in Oli-neu cells diminishes activation of both the PERK-eIF2α pathway and NF-κB in response to IFN-γ.This study also showed that activation of NF-κB in oligodendrocytes accompanies activation of the PERK-eIF2α pathway in transgenic mice that ectopically express IFN-γ in the CNS,and that enhancing IFN-γ-induced activation of the PERK-eIF2α pathway further increased NF-κB activation in oligodendrocytes (Lin et al., 2012). Moreover, Lin et al. generated a mouse model that allows for controllable activation of the PERK pathway specifically in oligodendrocytes. They found that PERK activation specifically in oligodendrocytes protects oligodendrocytes against inflammation during EAE, and the cytoprotective effects of PERK activation in oligodendrocytes are associated with NF-κB activation (Lin et al., 2013, 2014).Additionally, another study showed that activation of PKR(Protein kinase RNA-activated) can also result in NF-κB activation in oligodendrocytes through inhibition of IκBα translation (Tanner et al., 2015). Since IKKβ deletion in oligodendrocytes only blocks NF-κB activation through the IKKβ-dependent pathway, it is possible that IKKβ deletion alone is not sufficient to impair NF-κB activation in oligodendrocytes during EAE and to influence the disease development.
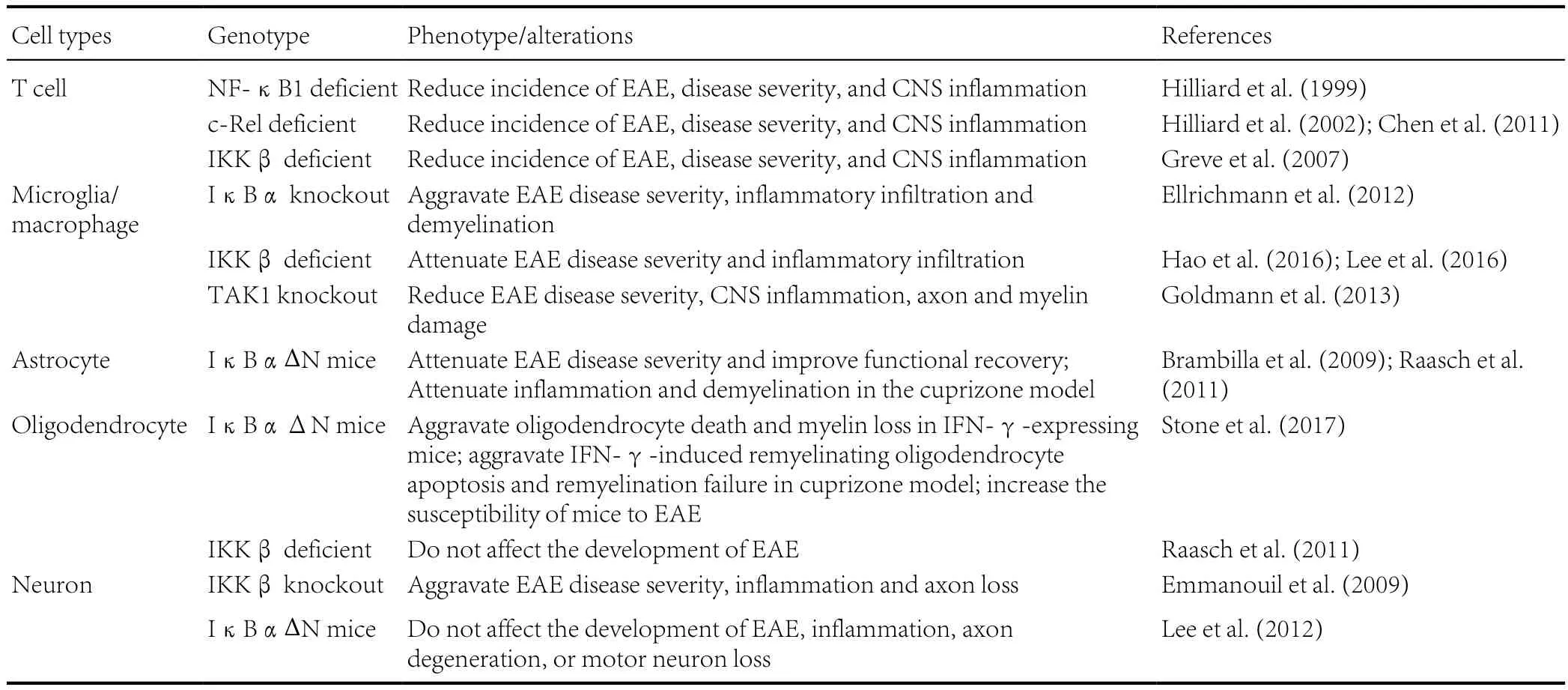
Table 1 Role of NF-κB in individual cell types in MS and EAE.
The Role of NF-κB Activation in Neurons in MS and EAE
A recent study suggested that neurodegeneration, particularly axon degeneration, is an early event and the primary cause of chronic disability in MS (Trapp and Nave, 2008). There is evidence to indicate that NF-κB activation in neurons regulates the morphology and plasticity of neurons, and is involved in learning and memory (Kaltschmidt et al., 2006; Kaltschmidt and Kaltschmidt, 2009). Emmanouil et al. generated a mouse model in which IKKβ is deleted from CaMKIIα (calcium/calmodulin-dependent kinase IIα)-expressing neurons using the Cre/lox system. Interestingly, deletion of IKKβ specifically in CaMKIIα-expressing neurons leads to a severe, non-resolving EAE, and increases inflammation and axon loss in the CNS of EAE mice, which is associated with the significantly reduced CNS production of neuroprotective factors and increased production of proinflammatory cytokine and chemokines (Emmanouil et al., 2009). These results suggest that NF-κB activation in CaMKIIα-expressing neurons is neuroprotective and has an inhibitory effect on inflammation in MS and EAE. On the other hand, Lee et al. generated a mouse model that expresses IκBαΔN, a super-suppressor of NF-κB, in CaMKIIα-expressing neurons using the tetracycline inducible system. In contrast, they found that expression of IκBαΔN in CaMKIIα neurons does not alter EAE disease severity, and does not affect inflammation, axon degeneration,or motor neuron loss in the CNS, suggesting that neuronal NF-κB signaling is not a major player in MS and EAE (Lee et al., 2012). Therefore, the role of NF-κB activation in neurons in MS and EAE remains elusive, and further investigations are required.
Conclusion
Evidence suggests that NF-κB activation in inflammatory cells, including T cells, microglia/macrophages and astrocytes,enhances inflammation and promotes the development of MS and EAE. Conversely, data indicate that NF-κB activation in oligodendrocytes and neurons protects these cells against inflammation in MS and EAE (Table 1). NF-κB exerts different roles in different cell types in MS and EAE, which are paradoxical and even conflicting. Due to this double-edged sword nature of NF-κB activation in MS and EAE, targeting NF-κB in MS may cause unpredictable side effects. Thus, it is unlikely that NF-κB is an ideal therapeutic target for MS.
Author contributions: YY, SS and WL wrote the manuscript.
Conflicts of interest: The authors declare no competing financial interests.Financial support: This study was supported by grants from the National Institutes of Health (NS094151 and NS105689) and the National Multiple Sclerosis Society (RG5239-A-3) (to WL).
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open access statement: This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike 4.0 License, which allows others to remix, tweak,and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewers: Manoj Kumar Jaiswal, Icahn School of Medicine at Mount Sinai, USA; Jiang Li, The Affiliated Hospital of Qingdao University, China.
Additional file: Open peer review reports 1, 2.

- 中国神经再生研究(英文版)的其它文章
- 3′-Daidzein sulfonate sodium protects against memory impairment and hippocampal damage caused by chronic cerebral hypoperfusion
- Neuroprotection by immunomodulatory agents in animal models of Parkinson’s disease
- Nanometer ultrastructural brain damage following low intensity primary blast wave exposure
- Altered leukocyte gene expression after traumatic spinal cord injury: clinical implications
- Promoting axonal regeneration following nerve surgery: a perspective on ultrasound treatment for nerve injuries
- Schwann cell Myc-interacting zinc-finger protein 1 without pox virus and zinc finger: epigenetic implications in a peripheral neuropathy