
唐氏综合征合并先天性心脏病相关基因的研究进展
2018-07-30 09:31周园,张斌
中国临床医学 2018年3期
周 园, 张 斌
复旦大学附属妇产科医院产科,上海 200011
唐氏综合征(Down syndrome,DS),又称21-三体综合征,其细胞遗传学基础是21号染色体呈三体性。约50%的DS患儿合并不同类型的先天性心脏病(congenital heart disease,CHD)[1-6]。
DS患儿合并的CHD的类型主要以间隔类缺损为主,包括房间隔缺损(atrialseptal defect,ASD)、室间隔缺损(ventricular septal defect,VSD)、房室间隔缺损(atrioventricular septal defect,AVSD)等。其中,完全性AVSD是DS特有的心脏畸形[7]。此外,还有部分DS患儿合并动脉导管未闭、肺动脉闭锁、右心室双流出道或复合缺损等心脏畸形[8]。DS的发病主要与卵母细胞减数分裂错误[9]、黏连蛋白异常[10]及端粒长度缩短[11]等有关;而CHD的发生与染色体非整倍体性、染色体上的微小缺失(如22q11.2缺失、7q11.23缺失等)、拷贝数变异(copy number variations,CNVs)及单核苷酸多态性(single nucleotide polymorphisms,SNPs)等相关。本文主要就DS合并CHD患儿的基因特点作一综述。
1 DS合并CHD的流行病学特点
DS合并CHD存在性别、种族的差异。Freeman等[12]通过对美国国家DS项目(national Down syndrome project,NDSP)和其前身亚特兰大DS项目(Atlanta Down syndrome project,ADSP)的数据进行分析,发现DS患儿合并的CHD中以AVSD为主,且呈现明显的性别差异,女婴的发生率约为男婴的2倍(优势比1.93,95%可信区间1.40~2.67);黑种人AVSD发生率高于白种人(优势比2.06,95%可信区间1.32~3.21);西班牙裔人群AVSD的发生率较低(优势比0.48,95%可信区间0.30~0.77)。此外,Diogenes等[13]也发现,DS女婴合并CHD的发生率明显高于男婴,CHD以间隔类缺损为主。关于DS合并CHD性别、种族差异的原因尚未明确,需要进一步探索。
2 DS合并CHD的2种假说
参与人类AVSD发生的相关基因有GATA4、Nkx2.5、TBX5、DSCAM、COL6 A1~2等[14],其中部分位于21号染色体上。DS合并CHD的发病机制在基因水平上主要有基因剂量效应假说和基因突变假说。
2.1 基因剂量效应假说 DS合并CHD的相关基因包括DSCAM、RCAN1、COL6 A1~2、CRELD1、ALK2及KCNJ6,其中DSCAM、RCAN1、COL6 A1~2及KCNJ6位于21号染色体上的关键区域21q22(图1)。DS的特征表型(如颅面部畸形、心血管系统畸形、智力减退等)与此区域相关[15-16]。21-三体引起基因量倍增效应,其候选基因呈现不同程度的表达增加,通过增加黏附分子的表达、干扰信号通路的调节等,导致心脏发育异常,形成CHD[17]。

图1 第21号染色体上与DS合并CHD有关的关键基因
2.2 基因突变假说 CRELD1及ALK2基因位于其他染色体上(图2),但在21-三体背景下,其上某些位点突变,从而导致CHD的发生,这是基因突变假说。然而,并不是所有DS患儿均患CHD,且患CHD的类型、程度各异,这可能是多基因作用的结果;21-三体可影响其他染色体上基因的表达,呈现基因的系统效应[18]。
3 基因特点
3.1 DSCAM基因 通过定量印迹剂量分析及荧光原位杂交技术对21号染色体进行分析发现,DS合并CHD发生的关键区域包含DSCAM、D21S55、ERG、ETS2等重要的基因位点,其中,DSCAM基因是已知的64个位于DS合并CHD关键区域中唯一调节细胞间黏附的基因[19]。DSCAM基因位于21号染色体长臂(21q22.3),其编码的跨膜蛋白在细胞黏附分子免疫球蛋白超家族中具有高度的序列和结构同源性,且其编码的蛋白鼠类和人类具有高度保守性。在鼠类心脏发育过程中,DSCAM基因在心内膜垫融合之前已高度表达。将人类DSCAM基因的cDNA转染至小鼠成纤维母细胞L细胞,DSCAM稳定的转化株表现出强大的黏附性能,DSCAM蛋白以亲同种抗原的方式快速形成聚合体[19]。
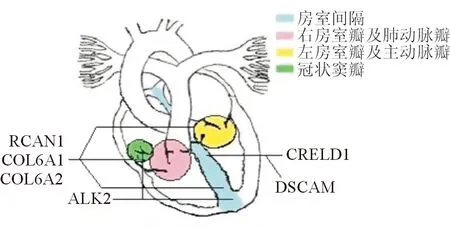
图2 DS合并CHD关键基因影响的心脏结构简图
黏连蛋白位于染色体着丝粒附近,在卵母细胞的减数分裂过程中,维持同源染色体的稳定性;其表达减少将导致非整倍体卵子的产生增加[20],进而导致DS的发生。而在DS患儿中,由于21号染色体三体化,位于其上的基因量倍增。这种剂量效应使细胞间黏连蛋白的表达在心内膜垫融合前异常增加,进而使细胞间黏附力增加,影响心内膜垫的融合,发生AVSD[19,21-22]。
3.2 RCAN1基因 RCAN1基因又称为唐氏综合征关键基因(Down syndrome critical region 1, DSCR1),位于人类21号染色体长臂(21q22.1),在DS合并CHD的发生中有重要意义[23]。利用DSCR1探针对包埋的胚胎进行RNA原位杂交,发现在胚胎发育第9.5~10.5天,以及之后的原始心管发育中(胚胎发育18~19 d起),尤其在动脉干、动脉球、原始心室及房室屏障的形成过程(胚胎发育18~19 d起)中高表达[23]。DSCR1基因编码钙调蛋白磷酸酶,负性调节钙调蛋白磷酸化。钙调磷酸酶可去磷酸化活化T细胞的核因子(nuclear factor of active T cells,NF-AT),而NF-AT是第1个被发现的只在心脏内皮细胞表达的转录因子,其过度表达可促进核异位及靶基因的激活。Ranger等[24]的实验发现,NF-AT基因靶向断裂的小鼠在胚胎发育13.5~17.5 d,心脏的形态发生出现明显异常,包括主动脉瓣和肺动脉瓣缺如,导致胎儿因充血性心衰而死亡[24-25]。因此,21-三体导致位于其上的DSCR1基因过度表达,使胚胎原始心管发育过程中钙调磷酸酶过度去磷酸化VF-AT,影响心脏瓣膜的正常发育及房室间隔的形成,导致先天性心脏发育缺陷。
3.3 COL6A1基因与COL6A2基因 COL6A1基因与COL6A2基因是编码胶原蛋白Ⅵ的2个基因簇,均位于21号染色体长臂(21q22.3)。胶原蛋白Ⅵ在胚胎第5~18周表达[26-28],参与原始房室间隔的形成,包括房室间隔瓣膜、房间隔中下部及膜部。DS患儿中,3倍的COL6A1基因与COL6A2基因破坏了胶原蛋白链表达的协同调节,进而影响DS患儿的胶原蛋白Ⅵ的表达[26]。另有学者指出[27],无论是否合并CHD,相比于染色体正常的对照组,21-三体心脏样本(包括心内膜垫的起源结构及瓣膜)中胶原蛋白Ⅵ的表达均明显增强,且这些组织发育明显不良。
3.4 CRELD1基因 CRELD1基因位于人类3号染色体短臂(3p25),是第1个被发现与人类AVSD相关的基因,其编码的蛋白质位于细胞膜表面,起类似细胞黏附分子的作用,参与正常心脏的发生。涉及CRELD1基因高度保守区域的突变可能是导致AVSD的原因,如C4201T突变及C4148T突变DS患儿中CRELD1基因某些保守位点的突变导致AVSD的发生[5-6, 29]。Maslen等[6]对39例DS合并AVSD患者的CRELD1基因进行测序,发现2例杂合错义突变,分别发生于cDNA外显子9第985位点(c.985C>T)及外显子10第1240位点(c.1240G>A)。这两个突变导致的相应的氨基酸替换(p.R329C、pE414K)在哺乳动物中高度保守,且均遗传自心脏发育正常的亲本,而在对照组(DS但心脏发育正常)未发现此突变。Li等[5]发现,Ts65Dn小鼠(经典DS模型小鼠)AVSD发生率为4.7%,Creld1+/-小鼠AVSD发生率为0%,而Creld1+/-Ts65Dn小鼠AVSD发生率上升至33.3%。
3.5 ALK2基因 骨形成蛋白(BMP)调节的信号通路参与胚胎心脏的分化和形成[30]。ALK2(activin-like kinase 2)是BMPs的Ⅰ型受体,位于1号染色体长臂。在发育中的小鼠胚胎心内膜结构上消除ALK2基因,可导致心内膜垫发育不全[31]。Smith等[32]对190例AVSD患儿的DNA标本进行筛查,发现2个位于ALK2编码基因上的SNPs(R307L、L343P),将ALK2基因上的SNPs之一(L343P)转染至斑马鱼胚胎,发现相较于野生型ALK2(wtALK2)斑马鱼胚胎,其BMP信号转导明显减少,并出现心管发育异常。在上述患儿中,发现其中1例合并原发孔型ASD及冠状窦分流异常,通过对其外周血淋巴细胞DNA测序,发现3个SNPs(ALK2 p.His286Asp、ALK3 p.Glu414Lys及ERBB3 p.Thr1169Ile)。其中,只有ALK2上的SNPs在2个实验样本中均导致蛋白质结构破坏,而在250例对照组中均未发现上述变化。家系分析发现,先证者父亲、母亲及姐姐分别携带上述1~2个SNPs,弟弟未携带上述SNPs,而超声心动图证实,以上亲属均未发生CHD,说明上述SNPs均不会在无21-三体的背景下单独导致CHD的发生。研究[33]得出相似结论。
3.6 KCNJ6基因 KCNJ6基因在DS患儿的非结构性心脏功能异常(如无症状的渐进房室传导阻滞及心动过缓等)中起重要作用。KCNJ6基因参与编码钾离子通道调节蛋白(G蛋白)的亚基-Kir3.2(GIRK2)。钾离子通道被G蛋白耦联受体激活,G蛋白耦联受体由异四聚体亚基(Kir3.1、Kir3.2、Kir3.3、Kir3.4)组成。其中,Kir3.2几乎表达于全身器官,包括心脏。钾离子通道激活后在窦房结形成负性变时效应,在迷走神经刺激下,心房收缩和房室传导减弱,从而导致心脏功能异常[34]。
4 小 结
目前已知的DS合并CHD的相关基因包括DSCAM、RCAN1、COL6 A1~2、CRELD1、ALK2及KCNJ6,其致病机制主要包括基因剂量效应及基因突变两种假说。DSCAM、RCAN1、COL6A1、COL6A2及KCNJ6位于21号染色体上,因21-三体导致其上的基因不同程度倍增,通过不同机制导致心脏发育异常,引起CHD。CRELD1及ALK2基因位于其他染色体上,因某些基因位点突变引起CHD。关于21-三体与上述DS合并CHD相关基因的相互作用机制须进一步的研究。此外,DS合并CHD发病与性别、种族的关系,以及黏连蛋白在DS的发病及DS合并CHD发病中的作用机制也值得进一步探讨。
猜你喜欢
华中科技大学学报(医学版)(2022年3期)2022-08-15
广州医药(2022年3期)2022-06-16
科学与社会(2021年3期)2021-12-02
协和医学杂志(2021年3期)2021-06-02
中国生殖健康(2020年7期)2021-01-18
中国生殖健康(2020年5期)2021-01-18
临床超声医学杂志(2020年3期)2020-03-30
中国生殖健康(2018年5期)2018-11-06
文学教育(2018年5期)2018-05-26
舰船科学技术(2016年1期)2016-02-27
