
CACNA1A基因突变导致发作性共济失调合并认知功能障碍1例家系报道
2018-07-24 07:41:56武强严华杨柳王伟莉舒雯欧冬梅
解放军医学杂志 2018年7期
武强,严华,杨柳,王伟莉,舒雯,欧冬梅
[作者单位] 430070 武汉 解放军武汉总医院神经内科(武强、杨柳、王伟莉、舒雯、欧冬梅),骨科(严华)
发作性共济失调(episodic ataxia,EA)是一种罕见的常染色体显性遗传病,近年来的基因连锁分析发现此类疾病是由于编码离子通道的基因突变所致。根据临床表现及基因定位EA可分为4型,其中,EA2型又称发作性共济失调伴眼球震颤(episodic ataxia,nystagmus-associated),或乙酰唑胺反应型周期性共济失调(acetazolamide-responsive hereditary paroxysmal cerebellar 2 ataxia,APCA),特征性表现为间歇性共济失调、构音障碍以及眩晕等。发作间期可见凝视诱发眼震,影像学检查中部分患者可出现小脑萎缩(蚓部显著)[1-2]。据国外文献报道,EA2型主要与钙通道基因(CACNA1A)突变有关[1],国内报道较少,且未见CACNA1A基因突变的EA2型合并认知功能障碍的文献报道。现报告1例EA2型合并认知功能障碍的临床特点。
1 资料与方法
1.1 临床资料 先证者,男,35岁,仓库保管员,10年前出现波动性行走不稳及吐词不清,间断发作头晕、飘浮感,每次持续半小时至数小时不等,近3年来自觉记忆力减退,易忘事、不能胜任目前工作。既往体健,无烟酒嗜好。查体显示构音不清,双眼侧视时可见水平粗大性眼震,双侧指鼻、跟膝胫试验欠稳准,闭目难立征阳性。脑磁共振(MRI)检查提示小脑萎缩,简易智能精神状态检查量表(MMSE)评测29分,蒙特利尔认知评估量表(MoCA)测评12分。动态心电图提示间歇性长R-R间期,最长2.2s,QRS波群形态改变,考虑早期复极综合征。
先证者二姑,女,48岁,退休工人,25年前出现发作性行走不稳及吐词不清,亦伴有发作性头晕、飘浮感,每次持续半小时可缓解。近5年自觉记忆力差,有时易迷路。既往体健。查体显示言语稍含糊,双眼侧视可见不持续水平眼震,余神经系统检查未见明显异常。脑MRI检查提示脑轻度萎缩,小脑为著,MMSE 20分,MoCA 8分。心电图正常。
先证者父亲,男,27岁结婚后每年发作性头晕,后出现行走不稳及言语含糊,但程度较轻。35岁左右记忆力减退,45岁后认知障碍逐渐加重,性格改变易暴躁。48岁疑心脏病猝死。无烟酒嗜好。
1.2 方法 获得先证者及其二姑知情同意后,抽取2人外周静脉血2ml,提取全血基因组DNA。检测受检样本SCA1、SCA2、SCA3、SCA6、SCA7、SCA8、SCA12、SCA17致病基因(CAG/CTG)重复序列区域。应用IDT xGen® Exome Research Panel进行目标区域捕获,使用illumina Hiseq X-ten二代测序仪对富集的文库进行PE150测序,平均测序深度为100X,95%以上的区域达到20X以上的覆盖度。测序后得到FASTQ格式的原始数据,通过软件确定短序列在基因组上的位置,进行排序和转换,寻找样本测序数据与参考基因组差异的位点(包括SNV和InDel)。使用gnomAD、ExAC、1000 Genomes Project、ESP6500等普通人公共数据库及In-House Database数据库过滤突变,对于常染色体显性或X连锁显性的变异位点,保留普通人数据库中频率小于0.01的变异;对于常染色体隐性或X连锁隐性的变异位点,保留普通人数据库中频率<0.05的变异。去除不影响剪接的同义变异位点,剩余的位于外显子及外显子附近20bp内的变异位点,结合SIFT、polyphen2等软件预测的有害性得分进行排序,再根据疾病表型及家系信息筛选得到可能的致病突变位点。在检测出的突变位点中进行Sanger验证排除假阳性,并在全部家系成员中验证是否出现共分离现象。
2 结 果
先证者及其二姑应用毛细管电泳基因分析受检样本SCA1、SCA2、SCA3、SCA6、SCA7、SCA8、SCA12、SCA17致病基因(CAG/CTG)重复序列区域未见异常扩增。利用靶向捕获-高通量测序方法,检测到先证者错义变异位点(CACNA1A基因的c.835C>T p.R279C变异,染色体位置chr19:13470563)。同时亦检测到先证者二姑存在一个相同错义变异位点(CACNA1A基因的c.835C>T p.R279C变异,染色体位置chr19:13470563)(图1)。
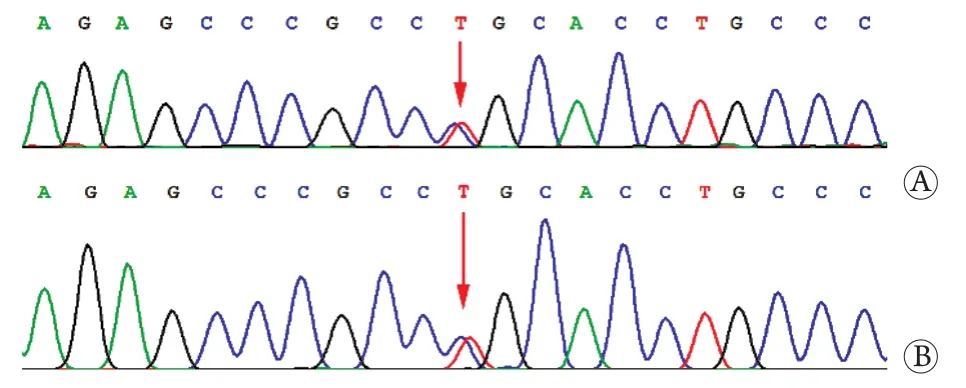
图1 该家系先证者及其二姑基因测序结果Fig.1 The results of Sanger sequence of the proband and his paternal aunt
查阅相关文献发现CACNA1A基因的c.835C>T p.R279C变异在ExAC普通人数据库东亚人群中的频率为0,生物信息学软件SIET和Polyphen2预测该变异分别为有害或可能有害变异,有文献报道该变异在多名EA 2型患者中检出,在一个已报道的家系中与疾病共分离[3-5]。基于以上证据,我们考虑CACNA1A基因的c.835C>T p.R279C变异为该家系的致病性突变。
3 讨 论
EA具有遗传异质性,为常染色体显性遗传,该病曾被称为急性短暂性普遍性小脑协调障碍,临床表现为发作性眩晕、共济失调和眼震,发作持续数秒或数周。运动、劳累、激素水平的变化和惊吓、姿势改变可诱发或加重发作[6]。EA是由编码离子通道的基因突变致病,其中EA2型致病基因定位于19p13,为CACNA1A基因突变引起。研究发现EA2型CACNA1A有许多突变形式:可有错义突变、截断突变、3端CAG重复扩增等。其中该基因突变导致转录后剪接或成熟前截断的改变,造成蛋白功能的丧失,是EA2的主要发病机制。
EA2型多在20岁前起病,可持续到成年。发作持续时间较EA1型长,可为数小时至数日。发作性主要特征是发作性共济失调、平衡障碍、构音障碍,50%以上的患者伴有眩晕恶心,约有一半患者出现偏头痛,约1/3患者出现特征性的自发性眼震,此外还可有耳鸣、眼睑下垂、复视,偶有振动幻觉。个别患者可有精神发育迟滞、癫痫发作。通常不伴有肌纤维颤搐。发作间期常有特征性的凝视诱发性眼震,下视、外视时尤为明显。有些患者后期可出现进行性小脑性共济失调,临床表现与脊髓小脑变性相似,后期可出现小脑萎缩。乙酰唑胺治疗EA2型一般有较好的疗效。本家系中共有3个家庭成员受累,临床均表现为发作性眩晕、共济失调及构音障碍,存活的2例中神经专科体检均见诱发性水平眼震,从临床表现上均符合EA2型。先证者及其二姑在发作间期仍有共济失调的体征并呈持续性,同时两人的影像学也均有小脑萎缩。考虑是由于这两人病程较长,已造成了小脑进行性的损害所致。本家系3例患者除了发作性共济失调外,后期均出现了不同程度的记忆力下降、视空间障碍等认知功能障碍,除先证者父亲猝死外,另两人客观认知测评包括MMSE、MoCA量表分值明显减低,临床上也出现不同程度的认知功能障碍。
上述特点与以往研究不同,也未检索到类似病例。我们考虑CACNA1A基因编码Cavl.2蛋白,形成P/Q型钙通道的孔道形成亚基,位于神经细胞膜上,此类通道广泛分布于脑部和神经肌肉接头处的神经末梢,主要功能是介导突触和神经末梢的递质释放。而Cavl.2蛋白是L型电压门控钙通道的主要组成部分,介导钙离子进入细胞内,在记忆形成、学习和行为中以及神经元存活、树突发育、突触可塑性等方面起着关键作用[7]。可能正是由于长期Cavl.2蛋白的异常,造成疾病后期认知功能障碍,但具体的发病机制还有待深入研究。另外本研究的家系中先证者父亲猝死,我们也查阅了相关文献,发现CACNA1A基因可能与以QT间期延长的间隔肥厚性心肌病、先天性心脏缺陷和心源性猝死为特征的心脏疾病有关。CACNA1A是长QT综合征的10种遗传相关基因之一[8],也有学者发现,在13例癫痫患者猝死的危险基因突变中,有4例检测到CACNA1A基因的突变[9]。因此,可认为先证者的父亲有可能也是因为CACNA1A基因突变引起的发作性共济失调并猝死。另外先证者动态心电图见间歇性长R-R间期、QRS波群形态改变,提示先证者存在心脏疾患,是否会出现心源性猝死还有待观察和随访。
我们报道的这个由CACNA1A基因突变所致的EA2家系中共有3个家庭成员受累,临床表现除了具有典型的发作性共济失调外,2例查体发现有典型的诱发性眼震,先证者心电图见间歇性长R-R间期,QRS波群形态改变,1例可疑心源性猝死,3例均合并有认知功能障碍。2例基因测序结果提示CACNA1A基因的c.835C>T p.R279C变异,本研究提示CACNA1A基因的c.835C>T p.R279C突变很可能是一种新型的合并认知功能障碍甚至猝死的EA 2型。
猜你喜欢
临床输血与检验(2022年3期)2022-06-22 02:52:50
海燕(2021年5期)2021-04-30 04:40:02
辽河(2020年1期)2020-02-12 05:11:53
安徽文学(2019年11期)2019-11-15 03:01:45
郑州大学学报(医学版)(2019年3期)2019-06-03 06:19:32
传染病信息(2019年2期)2019-05-17 13:16:04
广东海洋大学学报(2015年4期)2016-01-13 08:39:30
听力学及言语疾病杂志(2015年5期)2015-12-24 01:47:04
首都医科大学学报(2015年4期)2015-12-16 13:00:08
重庆医学(2015年12期)2015-03-05 05:52:54
