
炙甘草的质量评价
2018-07-11 09:43王莎莎蔡广知
中西医结合心血管病杂志(电子版) 2018年15期
王莎莎,赵 凌,蔡广知*
(长春中医药大学药学院,吉林 长春 130117)
1 介 绍
炙甘草为甘草的炮制加工品,即将甘草饮片。照蜜炙法(通则0213)炒至黄色至深黄色,不粘手时取出,晾凉。炙甘草性味甘,平。归心、肺、脾、胃经。具有补脾和胃,益气复脉的功效[1]。炙甘草主要含三萜类、二氢黄酮类及查尔酮类成分。蜜制后,甘草苷和甘草酸含量下降,同时产生醛糖衍生物等化学成分[2]。研究表明甘草素为甘草主要活性成分之一,具有抗癌[3]、预防糖尿病的应激反应[4]等活性,甘草素的提取分离及分析方法也日益受到关[5]。本实验通过对15批次炙甘草的性状特点进行描述,对显微特征、薄层进行鉴别,对水分、总灰分、酸不溶性灰分和含量进行测定,根据所得实验结果,对炙甘草质量进行质量分析。
2 方法与结果
2.1 实验材料
2.1.1 实验仪器
DHG-9070型电热恒温鼓风干燥箱(上海精宏实验设备有限公司);SHZ-D(Ⅲ)循环水式真空泵(巩义市予华仪器有限责任公司);安捷伦1100高效液相(安捷伦有限公司);KQ-50B型超声波清洗器(昆山市超声仪器有限公司)。
2.1.2 实验试剂
正丁醇、乙酸乙酯、甲酸、乙醇、浓硫酸(北京化工厂);冰乙酸(西陇化工股份有限公司);甲醇(Fisher Chemical);乙醚(天津天泰精细化学品有限公司)。
2.1.3 实验试药
样品1(内蒙,8 1 6 00 11 9 1);样品2(内蒙,817030931);样品3(内蒙,161201);样品4(内蒙,160601);样品5(内蒙,170522);样品6(内蒙,170201);样品7(内蒙,170521);样品8(甘肃,20161109);样品9(甘肃,20171101);样品10(甘肃,170512);样品11(甘肃,170905);样品12(甘肃,1709030);样品13(甘肃,170031);样品14(甘肃,171209);样品15(甘肃,180102)。
2.2 实验过程与结果
2.2.1 性状
性状考察,多数批次炙甘草为类圆形或椭圆形。外表皮灰棕色或红棕色,微光泽,纵皱纹,外皮松紧不一。横切面为黄色至深黄色,具形成层环,射线呈放射状。微黏性,焦香气,味甜。炙甘草直径在0.419~2.517 cm范围内,厚度在3.01~6.80 mm范围内。性状如图1-4所示。
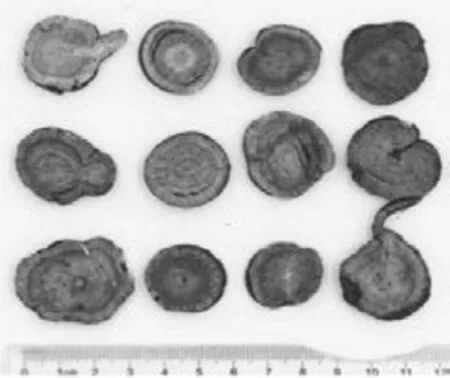
图1 炙甘草 甘肃 170702

图2 炙甘草 甘肃 20170608

图3 炙甘草 内蒙 161201

图4 炙甘草 内蒙 817030931
2.2.2 显微鉴别
观察可见纤维成束,壁厚,微木化,薄壁细胞含草酸钙方晶,晶鞘纤维。草酸钙方晶多见,呈长方形、类方形等形状。具缘纹孔导管较大,纹孔排列紧密,呈椭圆形。偶可见网纹导管,开口平而圆。木栓细胞多角形,红棕色,微木化。如图5-8。
2.2.3 薄层色谱鉴别
称炙甘草粉末(三号筛)约1 g,乙醚40 mL,回流1 h,过滤。30 mL甲醇重提,回流1 h,滤过蒸干,加水40 mL溶解。正丁醇提取3次,正丁醇液蒸干,5 mL甲醇溶解,作为供试品溶液。称甘草对照药材1 g,同法制成对照药材溶液。精密称甘草酸单铵盐对照品,制成每1 mL甲醇含2 mg的对照品溶液。如图9-11。
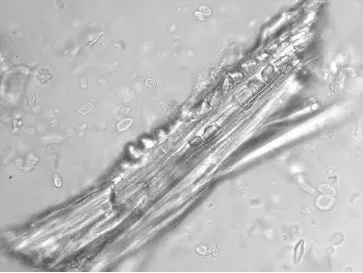
图5 炙甘草晶纤维
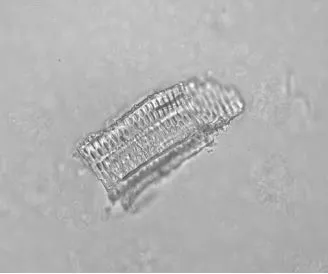
图6 炙甘草具缘纹孔导管

图9
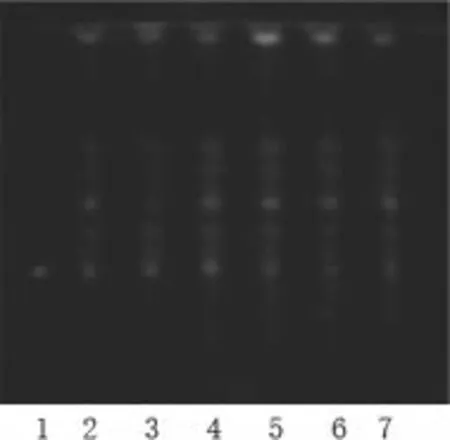
图1 0

图1 1

图7 炙甘草木栓细胞
图8 炙甘草草酸钙方晶
按2015版中国药典(通则0502)试验。乙酸乙酯、甲酸、冰醋酸、水=(15:1:1:2)为展开剂,展开,10%硫酸乙醇105℃下加热至斑点显色清晰,紫外光灯下检视结果。
如图可见,供试品的色谱中,对照药材色谱相应的位置上,会显现出颜色相同的荧光斑点;对照品色谱的相应位置上,可以看到相同的橙黄色的荧光斑点,符合2015版中国药典的标准。在相同点样量和展开的条件下,不同批次的药材薄层鉴别所显示的点颜色深浅有细微的差别。
2.2.4 水分测定
称炙甘草粉末(二号筛)2~5 g,平铺于扁形称量瓶。其余步骤按水分检查法(2015年版《中国药典》四部通则0832第二法)规定操作,根据减失重量,计算供试品的含水量(%)。

表1 水分测定结果(n,%)
如表可见,1 5批炙甘草样品中水分含量在6.22%~8.27%之间,2015版中国药典中规定的炙甘草水分≤10.0%,15批药材水分测定结果均合格。
2.2.5 灰分测定
2.2.5.1总灰分测定法
称炙甘草粉末(二号筛)2~3 g,放入坩埚。按灰分检查法(2015年版《中国药典》四部通则(2302))根据所得残渣的重量,计算供试品的总灰分的含量(%)。

表2 总灰分及酸不溶性灰分结果(n,%)
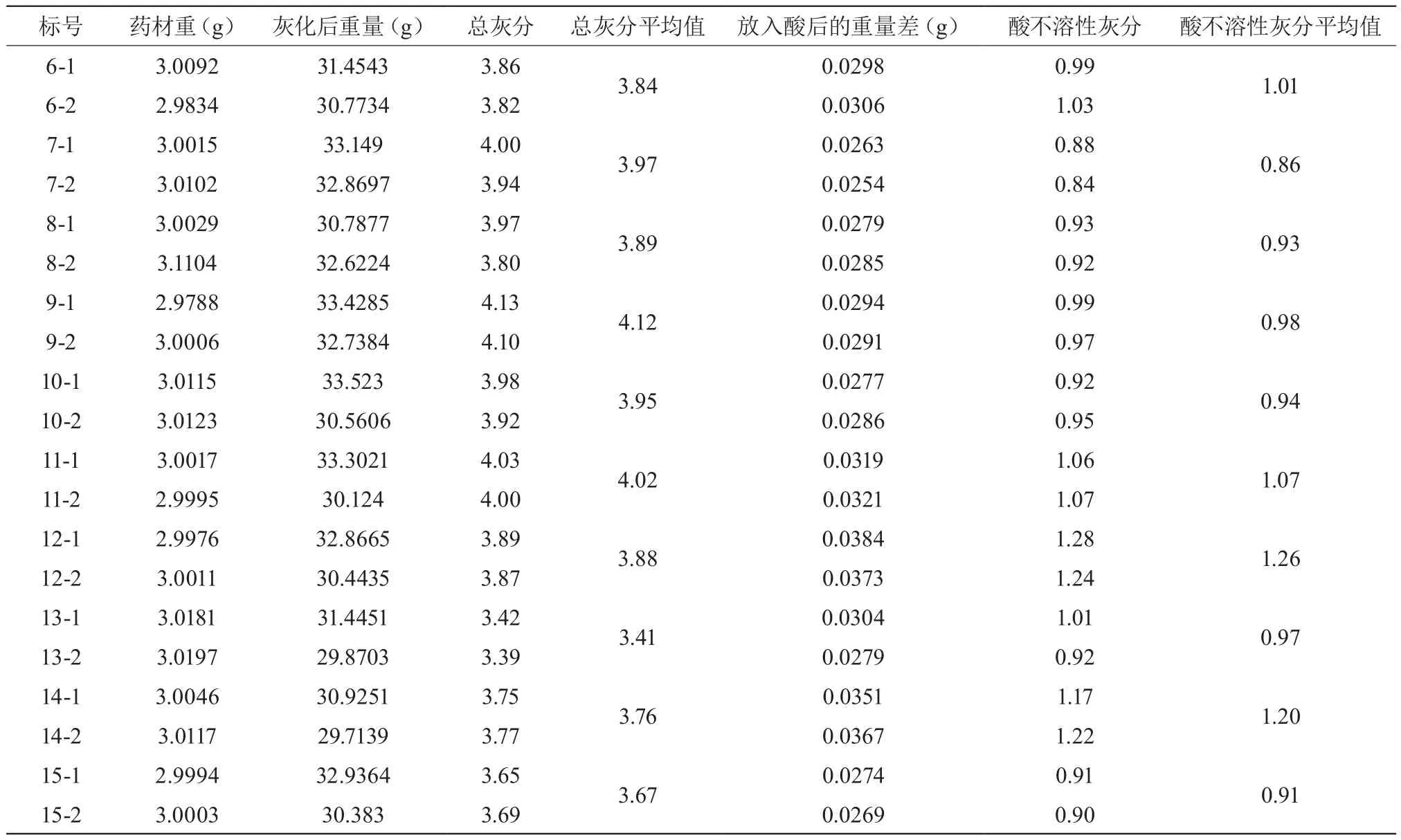
续表2
2.2.5.2酸不溶性灰分测定法
取2.2.5.1项下坩埚,加10 mL稀盐酸。参照2015年版《中国药典》四部通则(2302)规定操作,根据所得残渣的重量,计算供试品酸不溶性灰分的含量(%)。
数据见表2,结果显示,15批炙甘草药材的总灰分含量为3.41%~4.13%,酸不溶性灰分含量为0.86%~1.41%,符合2015版中国药典所要求得标准。
2.2.6 含量测定
参照2015年版《中国药典》(0512)测定。
2.2.6.1色谱条件与系统适用性试验
填充剂:十八烷基硅烷键合硅胶;流动相A:乙腈,流动相B:0.05%磷酸溶液;检测波长为237 nm。按照甘草苷峰理论板数计算,不低于5000。
2.2.6.2对照品溶液的制备
精密称甘草酸铵和甘草苷对照品。分别制成每1 mL70%乙醇试剂含甘草酸0.2 mg、甘草苷20 μg的溶液(甘草酸重量=甘草酸铵重量/1.0207)。
2.2.6.3供试品溶液的制备
精密称取炙甘草的粉末(过三号筛)约0.2 g,放到具塞的锥形瓶中,70%乙醇试剂100 mL,超声处理(功率250 W,频率40 kHz)30 min,冷却,称重,70%乙醇补足失重,滤过,取续滤液,即得。
2.2.6.4样品测定
分别称取15批炙甘草样品适量,按2.2.6.3项下的方法制备供试品溶液,按2.2.6.1项下的色谱条件测定。含量见图12-13。
药典规定,甘草酸不少于2.0%,甘草苷不少于0.5%。分析15批炙甘草得知,13批炙甘草含量明显不合格,仅2批炙甘草的含量合格,说明大部分市售炙甘草含量不符合2015版中国药典标准。
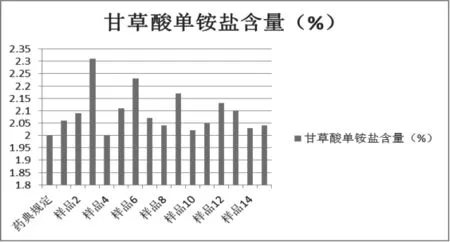
图1 2 炙甘草中甘草苷含量
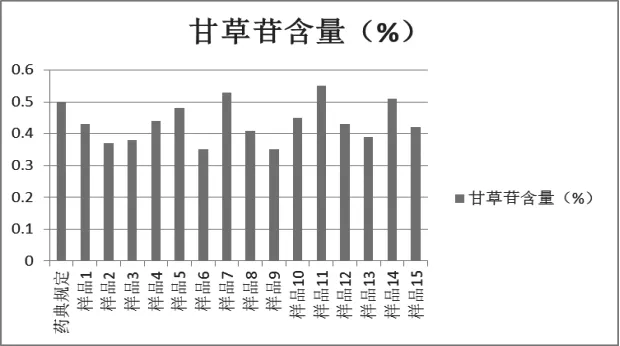
图1 3 炙甘草中甘草酸单铵盐含量
3 结 论
炙甘草质量评价的结果分析,13批炙甘草的含量不符合2015版中国药典标准,仅有2批含量较合格。根据结果可见,不同批次的炙甘草饮片的质量大部分为不合格产品,不符合2015版中国药典的标准,仅少部分炙甘草饮片的质量合格。原因可能有:1.购买的炙甘草饮片为各大药房购买的饮片,其贮藏环境和贮藏条件不符合贮藏要求,导致
炙甘草质量有所影响,贮藏时常常会因为湿度、温度、避光和空气流通等条件不当出现饮片质量下降的问题;2.炙甘草是甘草的炮制加工品,原药材含量不合格会直接导致其炮制加工品含量较低,甚至不符合药典标准,或者在炮制加工过程中净选、药用辅料、火候、翻炒时间等掌握不当出现炙甘草含量降低,导致市场流通的炙甘草大部分不合格。
猜你喜欢
中国药学药品知识仓库(2022年10期)2022-05-29
世界科学技术-中医药现代化(2021年5期)2021-11-05
中国药业(2021年12期)2021-06-28
中国合理用药探索(2021年2期)2021-01-03
中成药(2019年12期)2020-01-04
中医肿瘤学杂志(2019年2期)2019-08-15
新西部下半月(2019年6期)2019-08-11
中国中医急症(2019年10期)2019-05-21
科学与财富(2018年32期)2018-01-02
大观(2016年12期)2017-04-15
