
原发性干燥综合征患者合并肺动脉高压危险因素分析
2018-07-07 07:28李智腾范观止段培青张榕肖卫国
中国医科大学学报 2018年5期
李智腾,范观止,段培青,张榕,肖卫国
(中国医科大学附属第一医院风湿免疫科,沈阳 110001)
原发性干燥综合征 (primary Sjgren’s syndrome,pSS) 是一种以侵犯泪腺、唾液腺等外分泌腺体,具有淋巴细胞浸润和特异性自身抗体为特征的弥漫性结缔组织病,发病率为0.2%~1.4%,尤其好发于女性[1]。pSS的临床症状主要表现为口干、眼干,但约40%~50%的患者还可有其他的腺体外表现,可累及肺脏、肾脏、甲状腺及胆道系统等[2]。pSS患者肺脏受累多以间质性肺病为主要表现,而以肺血管受累为表现的肺动脉高压 (pulmonary arterial hypertension,PAH) 则是pSS的严重并发症之一[3],1年及3年生存率分别为73%和66%。因此,及早诊治PAH对pSS患者尤为重要。本研究拟回顾pSS合并与不合并PAH患者的临床资料并进行统计分析,旨在探究pSS患者发生PAH的危险因素。
1 材料与方法
1.1 研究对象
选取2014年至2016年于中国医科大学附属第一医院风湿免疫科住院的pSS相关PAH患者16例 (pSS-PAH组) 以及同时期所有初治的不合并PAH的pSS患者60例作为对照 (pSS-non-PAH组) 。其中,pSS-PAH组均为女性,年龄16~74岁,平均(50.13±15.67) 岁;pSS-non-PAH组亦均为女性,年龄26~80岁,平均 (52.90±14.5) 岁。2组年龄及性别无统计学差异 (P= 0.506) 。pSS相关PAH患者诊断符合2002年美国—欧洲共识组pSS分类标准或2012年美国风湿病学会提出的新的分类标准,并排除其他结缔组织病,如系统性红斑狼疮、系统性硬化病、类风湿关节炎等继发的干燥综合征。PAH诊断依据:以心脏彩超观测到肺动脉段增宽,三尖瓣返流,估测肺动脉收缩压 (pulmonary arterial systolic pressure,PASP) > 40 mmHg为标准,同时排除左向右分流的先天性心脏病、左心瓣膜病、肺栓塞、间质性肺病等导致PAH的其他疾病。
1.2 研究方法
收集2组的血常规、肝肾功、血清离子、心肌酶、凝血功能、免疫球蛋白、甲状腺功能、自身抗体等实验室检验指标及超声心动图相关数据等临床资料。
1.3 统计学分析
采用SPSS 17.0统计软件,对计数资料计算频率分布,采用χ2检验;对计量资料计算平均值和标准差,采用独立样本t检验 (符合正态分布) 或非参数检验的Mann-WhitneyU检验 (非正态分布) ;相关性分析采用Spearman非参数相关分析;对存在统计学差异且与PASP呈相关性的变量应用logistic回归进行危险因素分析。P< 0.05为差异有统计学意义。
2 结果
2.1 2组患者实验室数据的比较 (表1)
pSS-PAH组与pSS-non-PAH组比较,乳酸脱氢酶[ (349.86±253.30) U/L vs (217.44±165.17) U/L]、谷草转氨酶[ (41.50±34.37) U/L vs (24.61±18.68)U/L]、血清尿酸浓度[ (374.20±128.26) μ mol/L vs(292.13±244.64) μ mol/L]、磷离子浓度[ (1.30±0.13)mmol/L vs (1.16±0.18) mmol/L]水平均明显升高(P< 0.01,P< 0.05,P< 0.05,P< 0.01) ;而甲状腺激素T3水平[ (3.24±0.90) pmol/L vs (3.97±0.68) pmol/L]、血清镁离子浓度[ (0.82±0.09) mmol/L vs (0.88±0.07)mmol/L]降低 (P< 0.01,P< 0.05) 。另外,2组患者自身抗体阳性率均无明显统计学差异 (P> 0.05) 。
2.2 2组患者超声心动图相关指标比较 (表2)
在超声心动图的各项指标中,与pSS-non-PAH组比较,pSS-PAH组右心室内径[ (28.69±9.33) mm vs (18.58±2.10) mm]、肺动脉内径[ (28.19±5.09)mm vs (22.0±2.29) mm]水平明显升高 (P< 0.01,P< 0.01) ;而左心室舒张末期内径[ (40.50±6.70)mm vs (45.07±3.78) mm]、每搏量[ (40.75±6.90)mL vs (51.32±11.57) mL]、左心室收缩末期容积[(21.94±5.90) mL vs (29.51±7.89) mL]、左心室舒张末期容积[ (62.69±11.79) mL vs (80.76±17.93) mL]降低 (P< 0.05,P< 0.01,P< 0.01,P< 0.01) 。pSS-PAH组心包积液的阳性率较pSS-non-PAH组升高 (42.9% vs 5.4%,P= 0.001) 。
2.3 PASP与临床指标的相关性分析 (图1)
PASP与T3、血清镁离子浓度呈负相关(r=-0.343,P= 0.005;r= -0.256,P= 0.026) ,与磷离子浓度、乳酸脱氢酶、谷草转氨酶呈正相关 (r= 0.239,P= 0.038;r= 0.341,P= 0.004,r= 0.302,P= 0.008) ,PASP与左心室舒张末期内径、左心室收缩末期容积、左心室舒张末期容积、每搏量呈负相关 (r=-0.475,P< 0.01;r= -0.408,P< 0.01;r= -0.427,P<0.01;r= -0.393,P< 0.01) ;PASP与右心室内径、肺动脉内径呈正相关 (r= 0.790,P< 0.01;r= 0.722,P<0.01) ;PASP与血尿酸无相关关系。
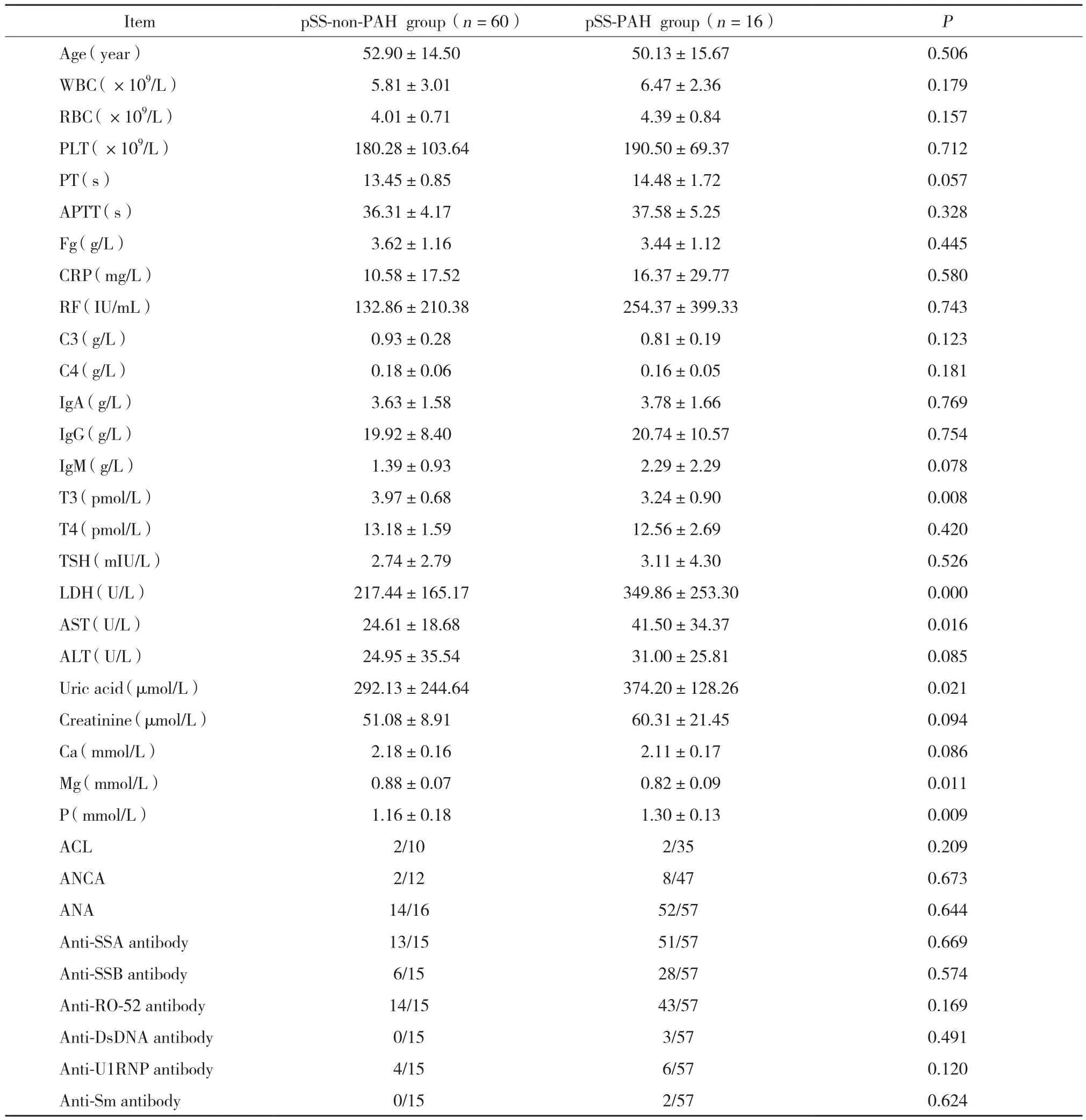
表1 pSS-PAH组与pSS-non-PAH组实验室检验结果比较Tab.1 Comparison of laboratory test results between the pSS-PAH and pSS-non-PAH groups
2.4 pSS患者合并PAH的危险因素分析 (表3)
排除肺动脉内径、右心室内径、左心室舒张末期内径等与PAH关系较明确的自变量,以pSS患者是否并发PAH为因变量 (有=1,无=0) ,以其他与PASP相关的变量为自变量,进行logistic回归分析。参与多因素logistic 回归分析的自变量有甲状腺素T3浓度、血清镁离子浓度、血清磷离子浓度、乳酸脱氢酶、谷草转氨酶。最后筛选出的有统计学意义的危险因素为甲状腺素T3浓度降低、血清镁离子浓度降低、血清磷离子浓度升高。
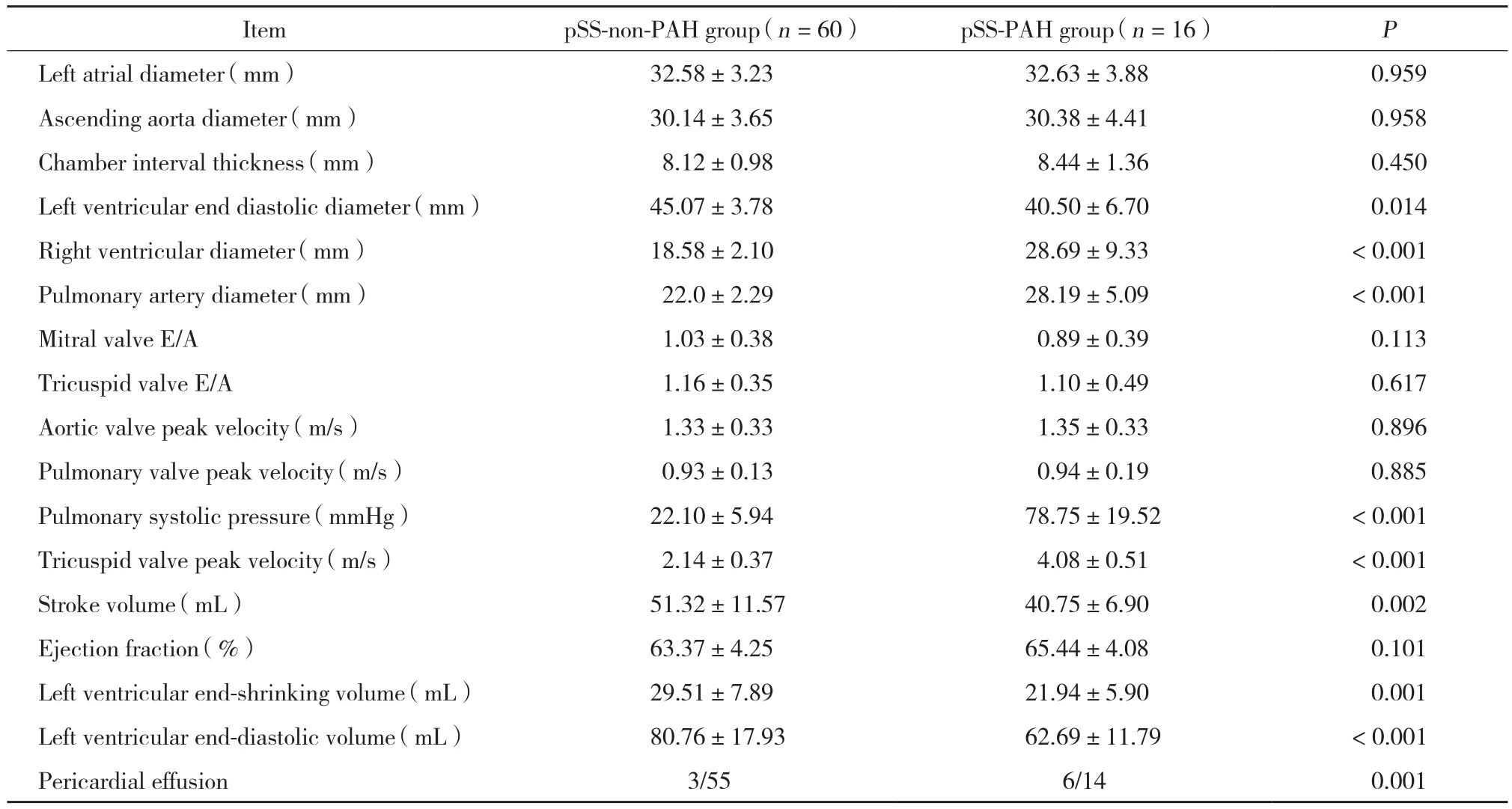
表2 pSS-PAH组与pSS-non-PAH组超声心动图结果比较Tab.2 Comparison of echocardiographic findings between the pSS-PAH and pSS-non-PAH groups
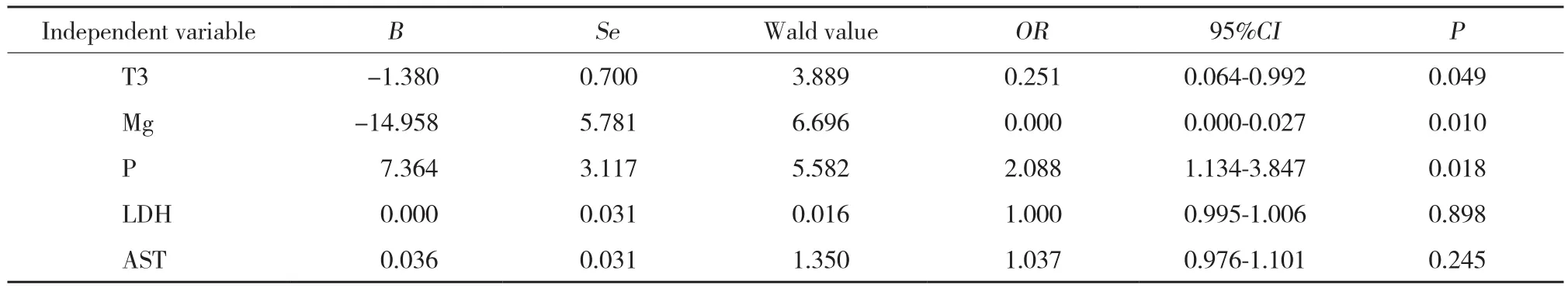
表3 pSS患者并发PAH的logistic回归分析Tab.3 Logistic regression analysis of PAH in patients with pSS
3 讨论
pSS相关PAH患者的预后不佳,积极研究pSS患者合并PAH的危险因素尤为重要。尽管已有多项结缔组织病相关PAH的危险因素研究,但多局限于系统性红斑狼疮及系统性硬化症,对pSS相关PAH的研究较少。本研究通过比较pSS-PAH及pSS-non-PAH 2组患者的实验室检验结果、超声心动图等指标,最后筛选出pSS相关PAH的危险因素为甲状腺激素T3浓度降低、血清镁离子浓度降低及血清磷离子浓度升高。乳酸脱氢酶、谷草转氨酶、右心室内径、肺动脉内径、左心室舒张末期内径、左心室收缩末期容积、左心室舒张容积、每搏量等指标在pSS-PAH及pSS-non-PAH 2组患者中也存在明显差异,且与PASP相关,这是由于PAH可造成右心功能不全,表现为心肌酶学异常;对心脏结构而言,肺动脉内径增宽、右心室内径增大、左心室舒张末期内径缩小、左心室舒张末期及收缩末期容积缩小等都是PAH导致的结果,而不是pSS患者并发PAH的危险因素。
血清T3水平和镁离子浓度降低及血清磷离子浓度升高与pSS患者合并PAH的机制是否相关?众所周知,甲状腺激素作为人体最重要的激素之一,能够促进新陈代谢,增加机体氧耗量。有研究[4]发现甲状腺素能增加血清儿茶酚胺浓度,导致肺血管收缩,血管顺应性下降,阻力增加;并通过增加前列环素、一氧化氮等舒血管物质的新陈代谢,减少5-羟色胺、内皮素1等缩血管物质的代谢,直接导致PAH形成。但本研究发现血清低T3水平与pSS-PAH相关可能与以下原因有关:一方面,在心肺血管平滑肌内,甲状腺素发挥生理作用不依赖于基因组活动,即不需要通过核内甲状腺素与甲状腺素受体结合启动相应的转录调节,而是通过激活细胞膜离子通道及内皮型一氧化氮氧合酶,导致血管平滑肌舒张,血管阻力及压力下降,从而增加心输出量[5];另一方面,甲状腺激素通过旁分泌方式增加邻近平滑肌细胞的内皮型一氧化氮产物促使血管舒张;因此甲状腺素减少可能造成心肺血管平滑肌收缩,外周血管阻力增加。BADESCH等[6]指出PAH与甲状腺功能减退多同时继发于自身免疫性疾病,而且CHU等[7]发现在PAH患者中,甲状腺自身抗体阳性率也显著高于非PAH患者;因此可以猜测两者或许有共同的自身免疫相关的病理生理机制,具体机制有待进一步研究。此外,有研究[3]发现PAH的发生与生长因子及炎症趋化因子的过度表达导致血管损伤明确相关,同时体内的炎性细胞因子又可在下丘脑、垂体或其他组织水平影响促甲状腺激素释放激素、促甲状腺激素、甲状腺素T3和甲状腺结合球蛋白的合成,并且降低细胞核T3受体的结合能力,从而使血清T3水平降低[8]。因此,笔者推测血清T3水平可能间接反映pSS患者炎症反应程度,进而间接提示PAH程度。甲状腺激素包括T3及T4,有研究[9-10]发现,T3作为机体甲状腺素的生理活性形式,发挥着一系列生理作用。而机体产生T4 (85%) 多于T3 (15%) ,通过骨骼肌、肝脏、肾脏等的5’-单脱碘酶1 (D1) 及5’-单脱碘酶2 (D2) 将T4转化为T3。但心肌细胞缺乏明显的单脱碘酶活动,因此心血管系统主要依赖血清T3水平发挥作用。
镁离子作为人体必需的矿物元素,对维持神经、肌肉及心脏应激至关重要。有文献[11-12]报道血清镁缺乏可导致炎症反应,激活巨噬细胞、中性粒细胞及其他免疫细胞,如白细胞介素6等;同时血清镁离子浓度降低可激活氧化应激或使抗氧化防御系统弱化,导致活性氧浓度升高,造成血管内皮损伤;YOSHIMURA等[13]发现镁离子作为钙离子的拮抗剂可使血管舒张。因此血清镁离子浓度降低可能通过促进炎症反应、氧化应激等使血管内皮细胞功能紊乱,造成血管重构,最终促进PAH的形成。
30%~50%的pSS患者可伴有肾小管损害,造成钙磷代谢障碍,导致体内血磷水平升高。本研究结果发现,血清磷离子浓度升高与pSS合并PAH风险增加相关。其原因可能与以下几方面有关:机体在磷离子浓度升高状态下,Na-P协同转运蛋白促使磷离子进入细胞,钙磷晶体沉积于血管壁造成血管钙化[14];在血管钙化过程中,血管平滑肌细胞分化或转化为成骨/软骨细胞,后者产生胞外胶原基质或非胶原蛋白的同时产生基质小泡,而一些富含钙、磷的外来体就附着于基质小泡,加速血管钙化[15];血清中过多的磷在血管壁沉积能损伤内皮细胞功能,导致一氧化氮产物减少,血管舒张功能障碍[16-19];磷离子浓度升高还能损伤内皮细胞的线粒体,诱导内皮细胞调亡,通过下调膜联蛋白Ⅱ降低内皮细胞生存力,并通过活化胰岛素样生长因子1受体及过度表达整合素连接激酶加速血管内皮细胞衰老。因此,血清磷离子浓度升高可能通过加速血管钙化,使血管顺应性下降及损伤血管内皮细胞数量与功能,加速血管炎的发生发展,促使血管阻力增加,最终导致PAH的形成。
综上所述,甲状腺素T3浓度与血清镁离子浓度降低或血清磷离子浓度升高可能参与pSS患者PAH的发生发展,如果pSS合并甲状腺素T3浓度降低、血清镁离子浓度降低或血清磷离子浓度升高中的任何一项,则应及早检查心脏超声或右心导管试验,尽早发现PAH,及时治疗,以获得良好的预后。
[1]DAFNI UG,TZIOUFAS AG,STAIKOS P,et al. Prevalence of Sjgren’s syndrome in a closed rural community [J]. Annal Rheumat Dis,1997,56 (9) :521-525.
[2]KREIDER M,HIGHLAND K. Pulmonary involvement in Sjgren syndrome.[J]. Semin Respir Crit Care Med,2014,35 (2) :489-500.DOI:10.1055/s-0034-1371529.
[3]LAUNAY D,HACHULLA E,HATRON PY,et al. Pulmonary arterial hypertension:a rare complication of primary Sjgren syndrome:report of 9 new cases and review of the literature [J]. Medicine,2007,86 (5) :299-315. DOI:10.1097/MD.0b013e3181579781.
[4]MARVISI M,BALZARINI L,MANCINI C,et al. Thyroid gland and pulmonary hypertension. What’s the link? [J]. Panminerva Med,2013,55 (1) :93-97.
[5]VARGASURICOECHEA H,SIERRATORRES CH. Thyroid hormones and the heart [J]. Horm Mol Biol Clin Invest,2014,18 (1) :15-26. DOI:10.1515/hmbci-2013-0059.
[6]BADESCH DB,WYNNE KM,BONVALLET S,et al. Hypothyroidism and primary pulmonary hypertension:an autoimmune pathogenetic link?[ J]. Ann Int Med,1993,119( 1) :44-46.
[7]CHU J W,KAO PN,FAUL JL,et al. High prevalence of autoimmune thyroid disease in pulmonary arterial hypertension[ J]. Chest,2002,122( 5) :1668-1673.
[8]WAJNER SM,GOEMANN IM,BUENO AL,et al. IL-6 promotes nonthyroidal illness syndrome by blocking thyroxine activation while promoting thyroid hormone inactivation in human cells[ J]. J Clin Invest,2011,121( 5) :1834-1845. DOI:10.1172/JCI44678.
[9]KLEIN I,DANZI S. Cardiovascular involvement in general medical conditions thyroid disease and the heart[ J]. Circulation,2007,116(15) :1725-1735. DOI:10.1161/CIRCULATIONAHA.106.678326.
[10]DAHL P,DANZI S,KLEIN I. Thyrotoxic cardiac disease[ J]. Curr Heart Fail Rep,2008,5( 3) :170-176.
[11]MALPUECHBRUGRE C,NOWACKI W,DAVEAU M,et al. Inflammatory response following acute magnesium deficiency in the rat[J]. Biochim Biophys Acta,2000,1501( 2/3) :91-98.
[12]WOLF FI,TRAPANI V,SIMONACCI M,et al. Magnesium deficiency and endothelial dysfunction:is oxidative stress involved?[ J].Magnes Res,2008,21( 21) :58-64.
[13]YOSHIMURA M,OSHIMA T,MATSUURA H,et al. Extracellular Mg2+inhibit scapacitative Ca2+entry in vascular smooth muscle cells[J]. Circulation,1997,95( 11) :2567-2572.
[14]TATSUMOTO N,YAMADA S,TOKUMOTO M,et al. Spironolactone ameliorates arterial medial calcification in uremic rats:the role of mineralocorticoid receptor signaling in vascular calcification[ J]. Am J Physiol Renal Physiol,2015,309( 11) :F967-F979. DOI:10.1152/ajprenal.00669.2014.
[15]CHEN NX,MOE SM. Pathophysiology of vascular calcification[ J].Curr Osteoporos Rep,2015,13( 6) :372-380. DOI:10.1007/s11914-015-0293-9.
[16]SHUTO E,TAKETANI Y,TANAKA R,et al. Dietary phosphorus acutely impairs endothelial function[ J]. J Am Soc Nephrol,2009,20(7) :1504-1512. DOI:10.1681/ASN.2008101106.
[17]DIMARCO GS,HAUSBERG M,HILLEBRAND U,et al. Increased inorganic phosphate induces human endothelial cell apoptosis in vitro[ J]. Am J Physiol Renal Physiol,2008,294( 6) :F1381-F387.DOI:10.1152/ajprenal.00003.2008.
[18]MARCO G SD,KNIG M,STOCK C,et al. High phosphate directly affects endothelial function by downregulating annexin Ⅱ[J]. Kidney Int,2013,83( 2) :213-222. DOI:10.1038/ki.2012.300.
[19]TROYANO N,NOGAL MD,MORA I,et al. Hyperphosphatemia induces cellular senescence in human aorta smooth muscle cells through integrin linked kinase( ILK) up-regulation[ J]. Mech Ageing Dev,2015,152:43-55. DOI:10.1016/j.mad.2015.10.001.
猜你喜欢
建筑与预算(2022年10期)2022-11-08
体育科技文献通报(2022年4期)2022-10-21
世界科学技术-中医药现代化(2022年3期)2022-08-22
中国临床医学影像杂志(2022年5期)2022-07-26
现代临床医学(2021年5期)2021-11-02
昆明医科大学学报(2021年6期)2021-07-31
现代临床医学(2021年2期)2021-03-29
心肺血管病杂志(2020年5期)2021-01-14
心肺血管病杂志(2020年5期)2021-01-14
哈尔滨轴承(2020年1期)2020-11-03
