
Ruboxistaurin对糖尿病大鼠心房重构的影响*
2018-06-29 08:06王海丽徐园园宫孟琦李健张跃程立君刘长乐刘彤李广平富华颖
中国心脏起搏与心电生理杂志 2018年3期
王海丽 徐园园 宫孟琦 李健 张跃 程立君 刘长乐 刘彤 李广平 富华颖
糖尿病是心房颤动(简称房颤)发生的独立危险因素[1]。糖尿病和房颤的发生密切相关。蛋白激酶C(PKC)是蛋白激酶ABC 家族的一部分,PKCβ属于经典PKC,依赖磷脂酰丝氨酸、Ca2+和甘油二酯或波脂激活。存在重复富含半胱氨酸C1结构域,氧化应激因素可以引起半胱氨酸残基的C1结构域氧化形成二硫键,被认为是过氧化或活性氧与PKC 激活的联系通路。
我们前期研究结果表明糖尿病心房肌中核转录因子-κB(NF-κB)p65表达上调,其下游产物肿瘤坏死因子-α(TNF-α) mRNA和转化生长因子-β(TGF-β)蛋白表达增多,导致心房重构。NF-κB活化和表达上调是导致心房重构发生的重要因素,但是引起心房NF-κB激活的上游机制尚未明确。
对大鼠腹腔巨噬细胞的研究表明,PKCβ能够引起NF-κB的激活以及诱导型一氧化氮合酶和TNF-α产生[2];在炎症细胞和心室肌细胞中PKCβ的激活可引起I-κB磷酸化,导致NF-κB活化[3];高血糖引起的NF-κB的激活和细胞因子IL-6的产生均可通过PKCβ的抑制剂逆转[4]。越来越多的证据表明PKCβ亚型在糖尿病并发症中异常表达和激活[5],在啮齿动物糖尿病心室肌细胞中PKCβ优先过表达或被激活,抑制PKCβ的活性能够改善糖尿病大鼠的心室功能[6-7]。糖尿病状态下心房肌的PKCβ是否被激活,其激活是否是NF-κB活化的上游机制还需要进一步研究明确。笔者采用PKCβ抑制剂RBX (Ruboxistaurinn,LY333531)干预,旨在探讨PKCβ通路在糖尿病大鼠心房重构中的作用及机制。
1 材料与方法
1.1材料
健康成年wistar雄性大鼠64只(北京华阜康生物科技股份有限公司),体重150~200g,进行两部分实验:第一部分为心脏超声测定后进行组织病理实验,第二部分为电生理实验,每部分32只大鼠。随机分为四组:对照组(control, C 组) ,糖尿病组(diabetes mellitus, DM 组),糖尿病+PKCβ抑制剂组(diabetes mellitus + RBX, DMR 组)和正常+ PKCβ抑制剂组(control + RBX, CR组),每组8只。DM组及DMR组大鼠以65 mg/kg[8]链脲菌素溶液腹腔一次性注射给药,C组及CR组以等容量的柠檬酸缓冲液腹腔一次性注射给药。CR 组及DMR组大鼠于造模成功后即予以RBX 1 mg·kg-1·d-1[9]灌胃8周,同时C组及DM组正常饮食8周。
1.2糖尿病模型的建立
链脲菌素(天津百倍生物)120 mg加入0.1 mmol/L柠檬酸钠缓冲液18 mL中,冰浴下充分溶解,过滤消毒,调节酸碱度使pH值为4.2。DM组及DMR组大鼠以65 mg/kg链脲菌素溶液腹腔一次性注射给药。72 h及1周时分别于清晨空腹(过夜禁食至少8 h,断尾取血)测量血糖,两次空腹血糖≥11 mmol/L或单次空腹血糖≥20 mmol/L表示造模成功。检测建模成功后第1、2、8周血糖及干预前、干预8周后体重并计算两者体重差值。
1.3心脏超声测定
CR组及DMR组大鼠于RBX灌胃前及灌胃8周后、C组及DM组同步进行心脏超声检查。将大鼠放入便携式多功能麻醉剂诱导箱中, 用异氟烷(吸入体积分数为2 %~3 %异氟烷和97 %~98 %的氧气)吸入麻醉后取出,并继续用异氟烷吸入维持麻醉,待麻醉稳定后,剪去前胸部及四肢体毛,连接体表心电图。应用超声仪(Visual Sonics Vevo2100成像系统),留取左室长轴观进行分析。于左室长轴观测量左房前后径(LAD)、左室舒张末内径(LVEDD)、左室收缩末内径(LVESD),测量3次取均值。干预8周后的两组大鼠于M型超声测量左室射血分数(LVEF),测量3次取均值。计算各组干预前与干预8周后LAD差值、LVEDD差值、LVESD差值。
1.4组织病理检查
1.4.1标本选取及处理 干预8周后且完成心脏超声测定后的各组大鼠,腹腔注射3%戊巴比妥钠50 mg/Kg麻醉。开胸,从主动脉根部离断心脏,放置于4℃PBS液中洗净残血,在冰台上取左房组织并固定于10%中性福尔马林液10~14 d。后进行乙醇梯度脱水、二甲苯透明、浸蜡等实验步骤制作组织蜡块并切片。每个标本取10张切片,每张切片厚3~4 μm[10]。
1.4.2HE染色 将制作好的组织切片经过脱蜡、脱水处理后用苏木精染液,伊红染液进行HE染色[10]。取HE染色切片,在400倍视野下选取包括多个细胞核位于中心且较圆的心肌细胞的横截面视野,每张切片选取10个细胞,测量心肌细胞横截面积,取均值。
1.4.3Masson染色 将制作好的组织切片经过脱蜡、脱水处理后用Weigiet铁苏木素、丽春红酸性品红、磷钼酸溶液、1%冰醋酸进行Masson染色[10]。取Masson染色切片,在400倍视野下,每张切片随机选取5个视野,图像以Tiff 格式存入电脑并应用Image Pro Plus 7.0软件计算胶原总面积及图像总面积。胶原容积分数(collagen volume fraction, CVF)=胶原总面积/图像总面积。
1.5电生理检查
用于电生理检查的各组大鼠于干预8周后开胸,于主动脉根部以上1 cm水平横断主动脉,取下心脏置于4℃台氏液中,冲洗并挤出心腔内残血后迅速将主动脉根部套扎于Langendorff灌流管上,灌流系统中温度为(37±0.5)℃的台氏液以100 cm H2O压力逆向灌流。将三对自制双极电极分别固定于右房( right atrium, RA)、左房(left atrium, LA)、右室(right ventricle, RV),极间距离2 mm(图1),将上述电极连接多道电生理记录仪(TOP-2001多道电生理系统,上海宏桐实业有限公司),同时记录出清晰的各部位双极电图。基础起搏周长(pace cycle length, PCL)设为150 ms。以周长150 ms起搏RA测定心房间传导时间(interatrial conduction time, IACT)。以周长150 ms开始,以5 ms递减测定房室传导文氏周长(Wenckbach cycle length of AV conduction, AVWCL)。以周长150 ms应用S1S2刺激法测定左房、右房有效不应期(atrial effective refractory period, AERP),并计算AERP的离散度(AERPD)。应用Burst刺激(起搏周长分别为50、40、30、25 ms)诱发房颤,每个起搏周长刺激5次,共刺激20次,每次间隔30 s。房颤定义为在Burst刺激后心房电图跟随着出现快速而不规则心房激动,同时心室呈不规则反应并持续超过1 s。

将三对自制双极电极分别固定于右房、左房、右室,极间距离2 mm
1.6统计学分析
2 结果
2.1四组一般状况、心脏超声指标比较
与C组相比,DM组及DMR组血糖均显著升高,体重均显著降低(P均<0.05);C组及CR组血糖、体重无差异(P均>0.05)(表1)。与C组相比,DM组干预8周后LAD及LAD差值显著增大,DMR组较DM组干预8周后LAD及LAD差值减小(P均<0.05);C组、CR组干预8周后LAD及LAD差值未见差异(P均>0.05);四组干预前LAD、LVEDD、LVESD,干预8周后LVEDD、LVESD、LVEF,LVEDD差值、LVESD差值均无差异(P均>0.05)(表2)。
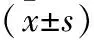
表1 四组血糖及体重比较
注:与C组比较,*p<0.05;与DM组比较,#P<0.05
表2 四组超声心动图参数
注:LAD=左房前后径;LVEDD=左室舒张末内径;LVESD=左室收缩末内径; LVEF=左室射血分数。 与C组比较,*P<0.05;与DM组比较,#P<0.05
2.2四组组织病理指标比较
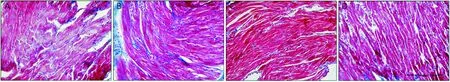
与C组相比,DM组大鼠左房肌细胞排列紊乱;与DM组相比,DMR组大鼠左房肌细胞排列整齐。与C组相比,DM组大鼠左房肌细胞横截面积增大[(86.67±21.95)μm2vs (42.75±18.36)μm2,P<0.05];与DM组相比,DMR组大鼠左房肌细胞横截面积减小[(48.36±16.85)μm2vs (86.67±21.95)μm2,P<0.05]。与C组相比,DM组大鼠左房肌胶原容积分数增大(5.64%±0.96% vs 3.00%±0.93%,P<0.05);与DM组相比,DMR组大鼠左房肌胶原容积分数减小(3.14%±0.80% vs 5.64%±0.96%,P<0.05);CR组及C组大鼠,左房肌细胞排列均整齐无差异,左房肌细胞横截面积大小无差异 (CR组:45.94±14.52μm2),左房肌胶原容积分数大小无差异 (CR组:2.23%±0.40%)(图2,图3)。

A:C组,B:DM组,C:CR组,D:DMR组
2.3四组电生理体征比较
与C组相比,DM组大鼠IACT延长, LAERP减小,RAERP减小,AERPD增大(P均<0.05);房颤诱发率升高(95/160 vs 10/160,P<0.05)。与DM组相比,DMR组大鼠IACT缩短,LAERP增大,RAERP增大,AERPD减小,P均<0.05;房颤诱发率明显减低(14/160 vs 95/160,P<0.05)。C组及CR组IACT, LAERP,RAERP,AERPD无差异(P均>0.05);C组及CR组房颤诱发率无差异 (CR组:12/160)(图4,表3)。
A:C组,B:DM组,C:CR组,D:DMR组

A:刺激前各部位双极电图,B:Burst刺激后双极电图
3 讨论
糖尿病是房颤发生的独立危险因素,而糖尿病引起房颤发生的机制目前尚不明确。糖尿病引起房颤的主要病理生理机制为结构重构及电重构。本研究结果表明PKCβ抑制剂RBX可以明显减轻糖尿病导致的心房肌细胞肥大及间质纤维化,并有效抑制糖尿病引起的AERP缩短、AERPD增大及IACT延迟等电重构,减少糖尿病大鼠房颤的诱发率。
表3 四组心率及PCL 150 ms时电生理参数比较
注:HR=心率;IACT=房间传导时间;AVWCL=房室传导的文氏周长;RAERP=右房有效不应期;LAERP=左房有效不应期; AERPD=心房有效不应期离散度。与C组比较,*P<0.05;与DM组比较,#P<0.05
越来越多的证据表明PKCβ亚型在糖尿病并发症中异常表达和激活[5]。在啮齿动物糖尿病心室肌细胞中PKCβ优先过表达或被激活,抑制PKCβ的活性能够改善糖尿病大鼠的心室功能[6-7]。链脲菌素诱导的糖尿病肾功能障碍的研究显示活性氧可激活PKC,丝裂原活化蛋白激酶(MAPK)和NF-kB,也可以上调TGF-β1,且肾小球系膜细胞通过晚期糖基化终末期产物蛋白的暴露也可激活PKCβ并增加TGF-β1的表达[11]。另外,用高糖孵化培养的心肌细胞是通过DAG-PKC信号通路促进NF-κB活化的,此过程增加的PKCα和PKCβ2使MAPK及随后的I-κB磷酸化[12]。高血糖状态下,可以通过PKCβ/NF-κB信号通路,增加TNF-α的mRNA表达,从而加重糖尿病炎症状态[13],而NF-κB活化过程的介质和产物(如TGF-β1等)能够引起炎症、内皮细胞功能失调、纤维化、心肌肥厚以及凋亡等,可能是造成糖尿病心房肌细胞肥大、心房肌纤维化等心房结构重构,进而促进房颤发生的原因。本研究结果显示应用RBX能够减轻心房的结构重构,可能是通过抑制PKCβ的激活,减少了其下游炎症因子如TGF-β1、TNF-α的表达。
糖尿病引起的心房间质纤维化,破坏了细胞间的偶联,阻碍了动作电位的传播,促进了心房折返环的形成。一些研究认为异常的心房电位、IACT延迟及AERP的缩短、AERPD增大等电生理特征是心房易损性增加的标志,是房颤发生的重要环节,与房颤的发生和维持有关[14]。此外动作电位时程的缩短及传导速度的减慢增加了房颤发生的可能。高糖引起的PKC激活可引起离子通道电流的改变,心房电重构可表现为心房肌细胞钙电流增大,细胞内钙超载,钠电流减小[15],细胞内钙超载可引起Na+/Ca2+交换,生成去极化电流,参与心肌细胞中钙波的发生和传播,引起延迟后除极,从而导致包括房颤在内的心律失常的发生。本研究结果表明RBX显著改善糖尿病导致的IACT延长、LAERP减小、RAERP减小、AERPD增大,减少房颤的发生。
阐明糖尿病导致房颤的上游机制是有效预防糖尿病导致房颤发生的关键,本研究结果表明RBX通过改善心房结构重构及电重构,减少房颤的发生,提示PKCβ激活可能在糖尿病引起房颤中发挥重要的作用,但研究结果尚需进一步深入的实验证实。
1 Movahed MR, Hashemzadeh M, Jamal MM. Diabetes mellitus is a strong, independent risk for atrial fibrillation and flutter in addition to other cardiovascular disease [J]. Int J Cardiol, 2005, 105: 315
2 Yu X, Zhang Q, Cui W, et al. Low molecular weight fucoidan alleviates cardiac dysfunction in diabetic Goto-Kakizaki rats by reducing oxidative stress and cardiomyocyte apoptosis[J]. J Diabetes Res, 2014, :420 929
3 Nagareddy PR, Soliman H, Lin G, et al. Selective inhibition of protein kinase C beta2 attenuates inducible nitric oxide synthase-mediated cardiovascular abnormalities in streptozotocin-induced diabetic rats[J]. Diabetes, 2009, 58: 2 355
4 Xia Z, Kuo KH, Nagareddy PR, et al. N-acetylcysteine attenuates PKCbeta2 overexpression and myocardial hypertrophy in streptozotocin-induced diabetic rats[J]. Cardiovasc Res, 2007, 73: 770
5 Lee WC, Chen HC, Wang CY, et al. Cilostazol ameliorates nephropathy in type 1 diabetic rats involving improvement in oxidative stress and regulation of TGF-β and NF-kB[J]. Biosci Biotechnol Biochem, 2010,74: 1 355
6 富华颖,李广平,刘彤,等.蛋白激酶C激活与糖尿病心房纤颤[J].中华糖尿病杂志,2013, 5(10): 634
7 Jang SM, Kim MJ, Cho MS, et al. Inhibitory effects of ursolic acid on hepatic polyol pathway and glucose production in streptozotocin-induced diabetic mice[J]. Metabolism,2010,59(4):512
8 Scivittaro V, Ganz MB, Weiss MF. AGEs induce oxidative stress and activate protein kinase C-(II) in neonatal mesangial cells[J]. Am J Physiol (Renal Physiol),2000,278:F676
9 Min W, Bin ZW, Quan ZB, et al. The signal transduction pathway of PKC/NF-kappa B/c-fos may be involved in the influence of high glucose on the cardiomyocytes of neonatal rats[J]. Cardiovasc Diabetol, 2009, 8:8
10 富华颖, 刘彤, 刘长乐,等. 普罗布考对糖尿病兔心房重构及心房颤动发生的干预作用[J].中华临床医师杂志, 2012, 6: 7 978
11 Ringvold HC, Khalil RA. Protein kinase C as regulator of vascular smooth muscle function and potential target in vascular disorders[J]. Adv Pharmacol,2017,78: 203
12 Wei L, Sun D, Yin Z, et al. A PKC-beta inhibitor protects against cardiac microvascular ischemia reperfusion in jury diabetic rats [J]. Apoptosis,2010,15(4):488
13 Pop-Busui R. Cardiac autonomic neuropathy in diabetes: a clinical perspective [J]. Diabetes Care, 2010, 33:434
14 McHugh D, Sharp EM, Scheuer T, et al. Inhibition of cardiac L-type calcium channels by protein kinase C phosphorylation of two sites in the N-terminal domain[J]. Proc Natl Acad Sci U S A, 2000, 97(22): 12 334
15 Liu C, Fu H, Li J, et al. Hyperglycemia aggravates atrial interstitial fibrosis, ionic remodeling and vulnerability to atrial fibrillation in diabetic rabbits[J]. Anadolu Kardiyol Derg, 2012, 12: 543
猜你喜欢
包头医学院学报(2022年5期)2023-01-04
临床超声医学杂志(2022年6期)2022-07-06
中西医结合心脑血管病杂志(2021年24期)2022-01-11
气象水文海洋仪器(2021年4期)2021-12-11
昆明医科大学学报(2021年5期)2021-07-22
临床超声医学杂志(2021年4期)2021-05-11
青岛大学学报(医学版)(2020年6期)2020-11-16
青岛大学学报(医学版)(2020年3期)2020-07-04
青岛大学学报(医学版)(2020年3期)2020-07-04
高中时代(2017年7期)2018-02-24
