
大肠杆菌中dctA基因敲除及其苹果酸摄取功能鉴定
2018-06-11 02:24:12姜巨全张正来
东北农业大学学报 2018年5期
姜巨全,张正来,徐 桐,孟 琳
(东北农业大学生命科学学院,哈尔滨 150030)
由于基因测序技术发展及其成本降低[1-2],目前公布的全基因组菌株逐渐增多。基因功能注释采用序列同源性比对方法完成[3]。该方法预测基因功能需相关试验鉴定。尽管体外蛋白功能分析是最直接手段,但很多基因仍需体内试验验证[4-5]。大肠杆菌缺失突变株功能互补被公认为体内基因功能分析最快捷、有效的手段之一,即基因敲除构建大肠杆菌目的基因缺失突变株,通过功能互补鉴定其他菌株相应基因是否具有类似功能。此外,大肠杆菌缺失突变株还可通过功能互补,对不具有同源性但具有相同功能新基因甚至功能未知基因作功能分析。如常使用大肠杆菌Na+/H+逆向转 运 蛋 白 基 因 缺 陷 株 KNabc(ΔnhaA,ΔnhaB,ΔchaA)[4]和EP432(ΔnhaA,ΔnhaB)[5]通过功能互补法,初步分析基因功能,从中度嗜盐菌[6-8]中筛选新型Na+/H+逆向转运蛋白基因[9-10]。
在大肠杆菌(E.coli)K12中,仅dctA基因编码1个有氧条件下负责摄取四碳-二羧酸的转运蛋白[11]。该蛋白能否摄取四碳-2-羟基羧酸(如苹果酸)未见报道。细菌中存在一类以Na+或H+作为共转运阳离子驱动柠檬酸、苹果酸或乳酸等2-羟基羧酸摄取的转运蛋白(2-hydroxycarboxylic acid transporter,2-HCT)[12]。
作为四碳-2-羟基羧酸代表,苹果酸常用作2-HCT转运蛋白功能鉴定底物。如果大肠杆菌(E.coli)K12 dctA基因具有摄取苹果酸功能,获得该基因敲除突变株,对其他菌株来源2-HCT转运蛋白基因克隆与功能分析具有重要意义。因此,本研究利用λ Red重组系统[13],通过基因敲除构建大肠杆菌四碳-二羧酸缺失突变株(E.coli ΔdctA),基于功能互补测试,鉴定DctA具有摄取苹果酸功能。该基因敲除突变株构建可为其他菌株来源2-HCT转运蛋白基因克隆与功能鉴定奠定基础。
1 材料与方法
1.1 材料
1.1.1 菌株、质粒和引物
主要菌株、质粒和引物如表1所示。
1.1.2 主要仪器
高速离心机(购自美国Thermo Scientific公司),超低温冰箱(购自美国Thermo Scientific公司),培养箱(购自上海志成生物公司),水浴锅(购自北京市永光明医疗仪器厂),电泳仪(购自北京六一生物科技有限公司),凝胶成像仪(购自美国SIM公司),pH计(购自德国Sartorius公司),高压灭菌锅(购自上海申安医疗器械厂),电转仪(购自德国Eppendorf公司),PCR仪(购自德国Eppendorf公司),分光光度计(购自天津市普瑞斯仪器有限公司)等。
1.1.3 主要试剂
200 bp DNA Ladder、 1 kb DNA Ladder、 Marker IV、RNaseA核糖核酸酶、IPTG(异丙基硫代-β-D-半乳糖苷))、X-gal(5-溴-4-氯-3-吲哚-β-D-半乳糖苷)购自天根生物公司。Eco R I购自TaKaRa生物公司。琼脂糖凝胶DNA胶回收试剂盒购自Omega公司。ATP、EasypfuDNAPolymerase、dNTP、TaqDNAPolymerase、pEASY T3克隆试剂盒以及Trans1-T1感受态细胞购自北京全式金生物公司。氨苄青霉素、氯霉素、L-阿拉伯糖、L-苹果酸和葡萄糖购自Biotopped公司。试验所需引物合成与测序,均由北京华大基因公司完成。
1.1.5 培养基及诱导剂母液
LB培养基(1 L):
胰蛋白胨10 g,酵母粉5 g,NaCl 10 g,pH 7,定容至1 L。
M9培养基:
① 5×M9培养基母液(200 mL):Na2HPO4·7H2O 12.8 g;NaCl 0.5 g;NH4Cl 1 g;KH2PO43 g,115℃灭菌30 min;
②含0.2%葡萄糖M9培养基(1 L):5×M9盐溶液200 mL;1 mol·L-1MgSO42 mL;1 mol·L-1CaCl20.1 mL;20%葡萄糖20 mL;无菌水定容至1 L。
③含0.2%苹果酸M9培养基(1 L):5×M9盐溶液200 mL;1 mol·L-1MgSO42 mL;1 mol·L-1CaCl20.1 mL;20%苹果酸20 mL;无菌水定容至1 L。
M9培养基配制过程中,MgSO4、CaCl2、葡萄糖、苹果酸均溶解于ddH2O,使用0.22 μm滤膜过滤除菌。按需求向培养基中添加终浓度为50 μg· mL-1氨苄青霉素或25 μg· mL-1氯霉素。
L-阿拉伯糖母液(4 mol·L-1):15.013 g·L-1阿拉伯糖溶解于ddH2O,定容至25 mL,使用0.22 μm滤膜过滤除菌。工作浓度为100 m·mol·L-1。
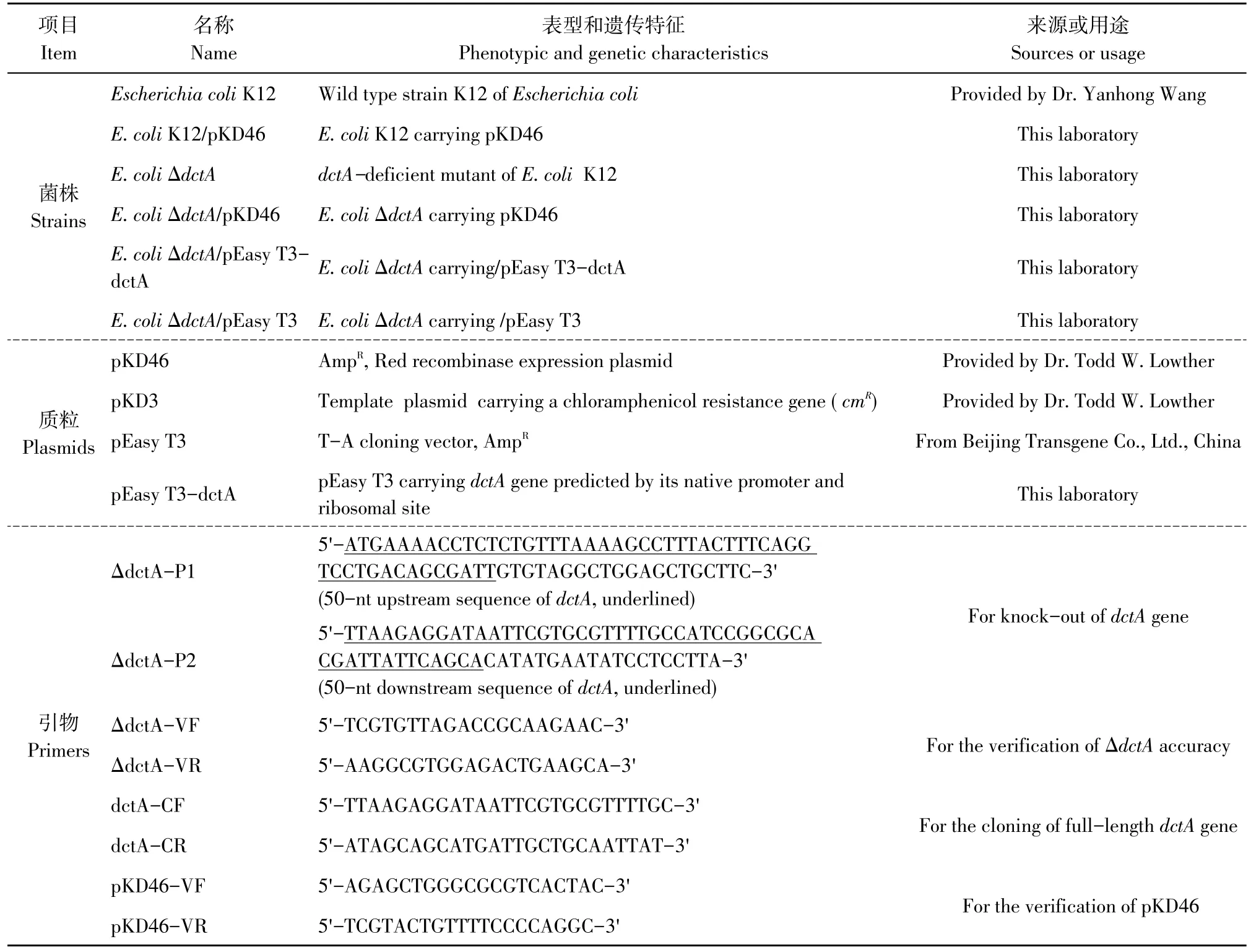
表1 菌株、质粒和引物Table 1 Strains,plasmids and primers
1.2 方法
1.2.1 含氯霉素抗性基因(cmR)及dctA同源臂PCR扩增
以质粒pKD3为模板,使用带有50 nt的dctA敲除引物(ΔdctA-P1和ΔdctA-P2,见表1),对氯霉素抗性基因(cmR)作PCR扩增。引物设计原则为保证以上引物5'端分别引入50 nt dctA基因序列作为该基因同源臂(见表1)。扩增条件见表2,退火温度为56℃,取1 μL PCR产物作0.7%琼脂糖凝胶电泳检测。该PCR产物即为含氯霉素抗性基因(cmR)及dctA同源臂目的片段。为验证其正确性,在PCR产物末端加“A”,反应体系为PCR产物中加入1 μL Taq DNA Polymerase、10×Buffer 5 μL 和 dNTP 4 μL,72 ℃扩增60 min。胶回收PCR产物2~4 μL,与0.5 μL pEASY T3载体混匀,室温放置约20 min,转化Trans1-T1感受态细胞,在含有Amp50和Cm25LB平板上作蓝白斑筛选,质粒提取后经酶切鉴定正确,送华大基因公司测序鉴定PCR产物。

表2 含cmR基因及dctA同源臂PCR扩增反应体系Table 2 PCR reaction system of cmRgene fragment flanked by dctA homologous arms
1.2.2 λ Red重组系统诱导表达及其电转化感受态细胞制备
将pKD46首先采用CaCl2法[14]化转大肠杆菌(E.coli)K12感受态细胞,挑取单菌落,接种于Amp505 mL LB液体培养基中,30℃,160 r·min-1过夜培养。以1%接种量转接于100 mL含Amp50的新鲜LB液体培养基,培养约2 h至OD600nm0.2~0.3;加入过滤灭菌L-阿拉伯糖(终浓度100 m·mol·L-1),30℃,160 r· min-1诱导培养1.5~2 h,至OD600nm约0.8时停止培养,立即冰浴20 min;分装菌液于预冷离心管中,离心收集菌体,无菌去离子水重悬洗涤1次;再用冰冷的10%甘油(去离子水配制)重悬、洗涤
2次;适量预冷10%甘油重悬收集菌体(比例为100 mL菌液:1 mL 10%甘油),按每管100 μL分装,直接用于电转化(离心参数为4℃,5 000 r·min-1和10min)。
1.2.3 含氯霉素抗性基因(cmR)及dctA同源臂电转化
将含氯霉素抗性基因(cmR)及dctA同源臂PCR产物(约400 ng)加入新制备100 μL感受态细胞中,混匀,转移至预冷0.1 cm电转杯中,BioRad电转仪作电转化(条件为:电压2 500 kV,时间4~5 ms)。电转后立即加入900 μL液体LB培养基复苏,混匀后转入无菌1.5 mL离心管中,37℃,160 r·min-1复苏1 h。然后,室温放置约12 h涂布于含25 μg·mL-1氯霉素LB平板上37℃培养16~24 h,直至菌落出现。
1.2.4 dctA基因敲除与验证
将氯霉素抗性平板长出单菌接种于含25 μg·mL-1氯霉素的LB液体培养基,37℃震荡培养。以上述菌液为模板(以E.coli K12/pKD46菌液作为负对照),使用ΔdctA验证引物(ΔdctA-VF和ΔdctAVR,见表1),作PCR扩增。扩增条件如表2所示,其中退火温度为56℃。PCR产物经0.7%琼脂糖凝胶电泳验证后,送北京华大基因公司测序鉴定基因敲除是否正确。
1.2.5 质粒pKD46消除与验证
质粒pKD46复制起点对温度敏感,该质粒可在37℃条件下正常复制,但42℃无法复制。为消除pKD46,将以上重组菌株划线于无抗生素LB平板,经42℃过夜培养后,牙签挑取单菌落分别于含有Cm25和Amp50液体LB培养基中,培养约12 h,观察生长情况。如果菌株在Amp50液体LB培养基生长,则重复以上步骤,直至Amp50液体LB培养基不再生长,暗示质粒pKD46已消除。在确定单菌落无法生长基础上,以质粒pKD46为对照,使用pKD46验证引物(pKD46-VF和pKD46-VR,见表1)对以上单菌落作PCR验证。扩增条件见表2,其中退火温度为55℃。
1.2.6 dctA基因克隆与载体构建
以大肠杆菌(E.coli)K12基因组为模板,使用dctA克隆引物(dctA-CF和dctA-CR,见表1),对dctA基因及上游启动子和核糖体结合位点作PCR扩增,扩增条件如表1所示,其中退火温度为55℃,取1 μL PCR产物作0.7%琼脂糖凝胶电泳检测。PCR产物末端加“A”、构建载体等同1.2.1方法所述。在含Amp50的LB固体培养基中蓝白斑筛选,质粒提取后经酶切鉴定正确,送测序鉴定载体构建是否正确。
1.2.7 苹果酸摄取功能互补测试
将野生型大肠杆菌(E.coli)K12及其基因敲除突变株,经LB培养基活化,以1%接种量转接于含0.2%葡萄糖作为唯一碳源M9液体培养基,培养至适宜OD600nm,去除LB培养基中痕量苹果酸;转接含0.2%苹果酸作为唯一碳源M9液体培养基,37℃、140 r·min-1培养72 h,绘制测试菌株生长曲线;基于以上结果,选取适宜生长时间,测试所有受试菌株OD600nm,验证受试菌株是否具有摄取苹果酸功能。
2 结果与分析
2.1 dctA基因敲除片段制备
为敲除大肠杆菌(E.coli)K12的dctA基因,采用1.2.1方法制备该基因敲除线性片段。在PCR扩增氯霉素抗性基因同时,引入50 nt dctA基因同源臂用于后续基因同源重组(原理见图1)。最终获得PCR产物长度为1 115 bp(见图2A)。该PCR产物即为dctA基因敲除线性片段,命名为ΔdctA-P1-CmR-ΔdctA-P2。为验证PCR产物正确性,将部分该PCR产物构建到pEasy T3。获得重组载体pEasy T3-ΔdctA经酶切验证(见图2B)正确后,进一步测序证明ΔdctA-P1-CmR-ΔdctA-P2制备成功。
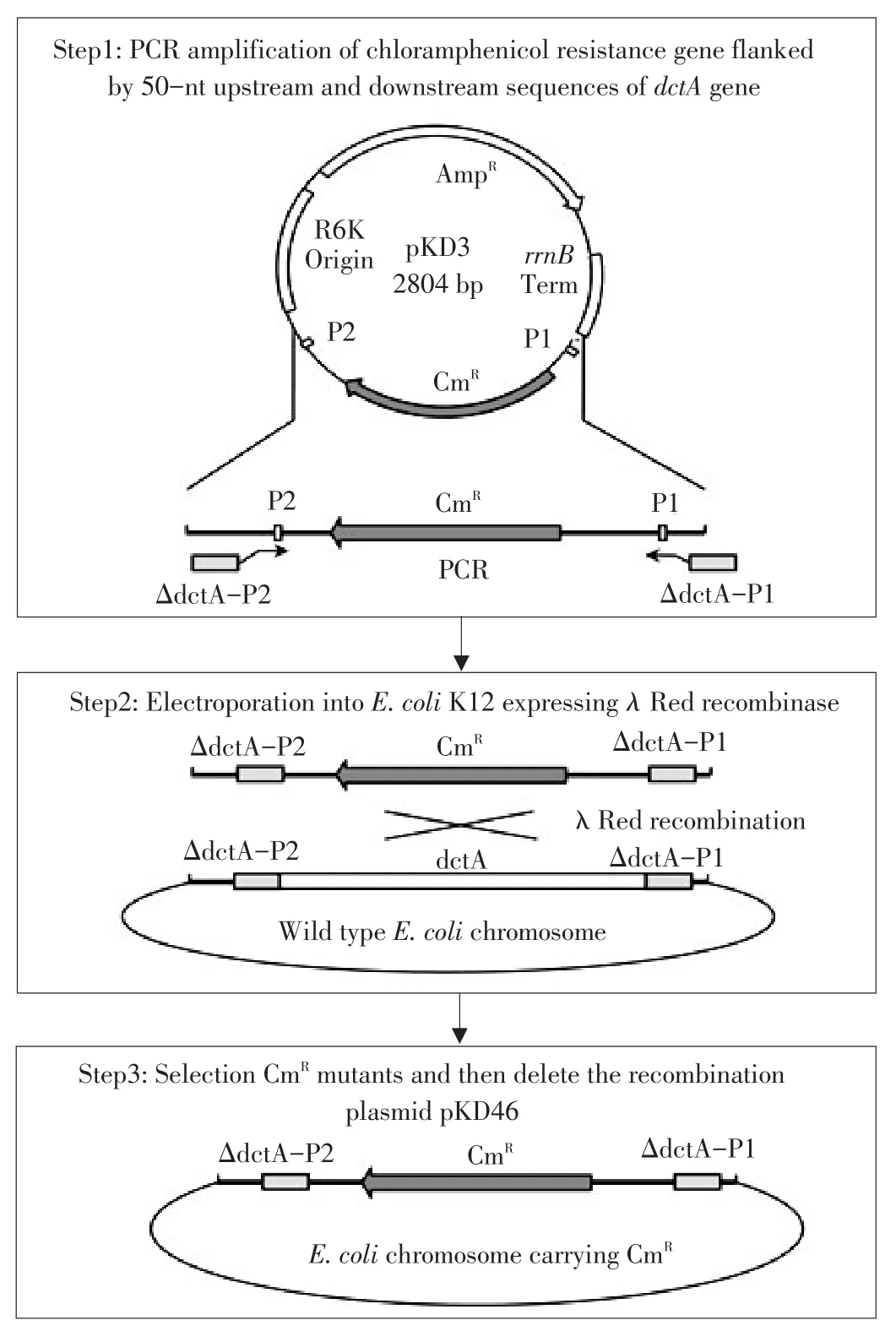
图1 λ Red同源重组示意图Fig.1 λ Red homologous recombination diagram
2.2 重组菌株E.coli ΔdctA/pKD46构建与验证
为构建dctA基因敲除突变株,将携带λ Red重组系统的pKD46化转大肠杆菌(E.coli)K12,诱导λ Red重组系统表达,制备E.coli K12/pKD46电转化感受态细胞;采用1.2.3方法将上述制备dctA基因敲除线性片段(ΔdctA-P1-CmR-ΔdctA-P2)电转化至E.coli K12/pKD46感受态细胞中,诱导ΔdctA-P1-CmR-ΔdctA-P2与野生型E.coli K12 dctA基因同源重组。随机挑取3个单菌落,采用1.2.4方法以E.coli K12/pKD46菌液作负对照,对以上3个单菌落PCR扩增,验证是否获得突变株E.coli ΔdctA。确定以上单菌落PCR产物小于E.coli K12/pKD46菌液PCR产物(见图3A)后,选取其一经测序,大肠杆菌(E.coli)K12基因组dctA基因已被ΔdctA-P1-CmR-ΔdctA-P2替代。表明dctA基因已成功敲除,该重组菌株命名为E.coli ΔdctA/pKD46。

图2 PCR扩增dctA基因敲除线性片段Fig.2 PCR amplification of linear fragment for knock-out of dctA gene
2.3 基因敲除突变株E.coli ΔdctA构建
为构建基因敲除突变株E.coli ΔdctA,需消除重组菌株E.coli ΔdctA/pKD46中质粒pKD46。因此,从双抗(Cm25和Amp50)LB平板挑取2个单菌落,采用1.2.5方法反复转接于无抗生素的LB固体平板,经42℃过夜培养后,最终在含Amp50液体LB培养基均不再生长,表明质粒pKD46已消除。在此基础上,以质粒pKD46为对照,经PCR验证,以上2个菌落无特征条带,表明质粒pKD46已消除(见图3B)。选取其一作为dctA基因敲除突变株用于后续功能验证,命名为E.coli ΔdctA。
2.4 dctA基因克隆及其表达载体构建
为进一步验证E.coli ΔdctA构建及DctA摄取苹果酸功能,需要构建pEasy T3-dctA表达载体。首先对dctA基因及上游启动子和核糖体结合位点(1 660 bp)作PCR扩增,PCR产物约为1.6 kb(见图4A),与实际大小基本一致。将PCR产物构建到pEasy T3,提取质粒经酶切验证(见图4B)后,测序结果表明pEasy T3-dctA表达载体构建成功。将pEasy T3-dctA(pEasy T3为负对照)转化E.coli ΔdctA感受态细胞,获得阳性克隆分别命名为E.coli ΔdctA/pEasy T3-dctA和E.coli ΔdctA/pEasy T3。
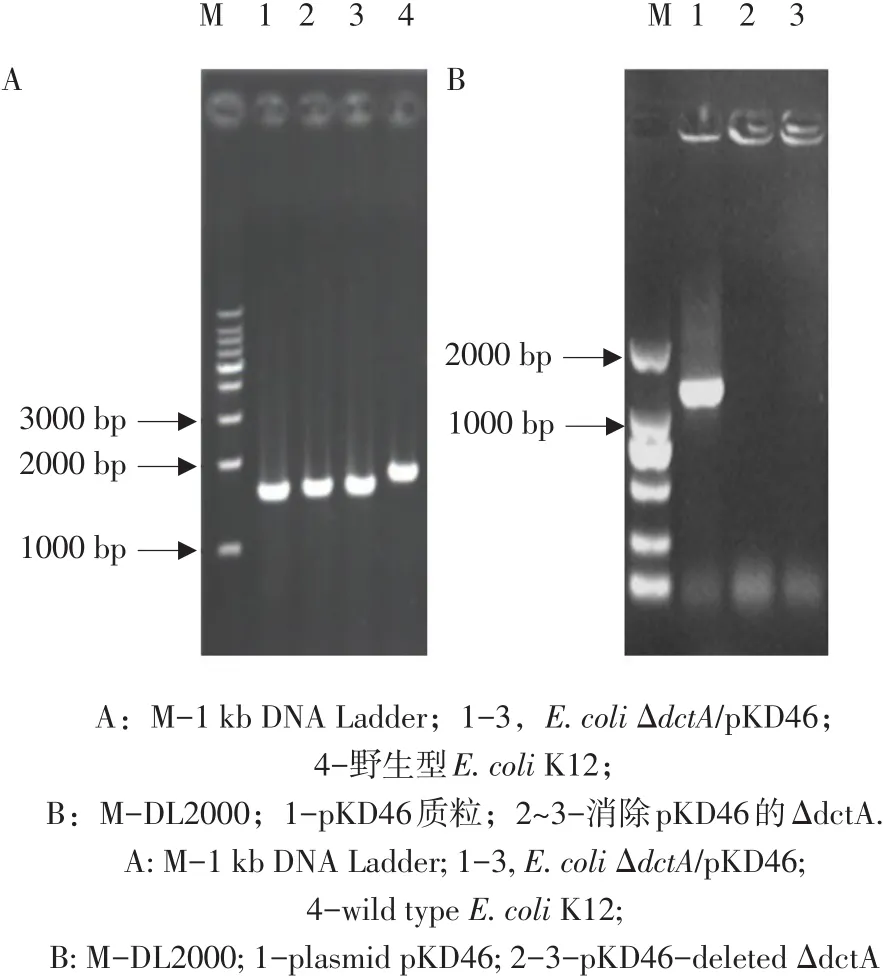
图3 基因敲除突变株E.coli ΔdctAPCR验证Fig.3 Verification of E.coli ΔdctA by PCR
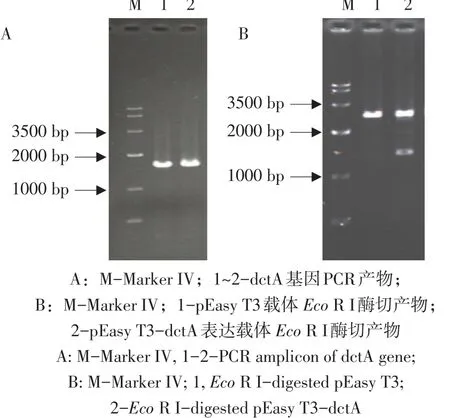
图4 pEasy T3-dctA重组载体构建Fig.4 Construction of recombinant plasmid pEasy T3-dctA
2.5 E.coli ΔdctA苹果酸摄取功能互补测试
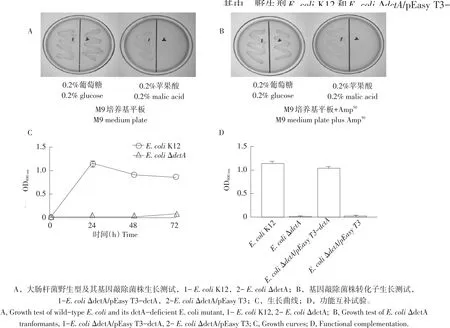
图5 基因敲除菌株E.coli ΔdctA功能互补试验Fig.5 Functional complementary experiment of E.coli ΔdctA
为测试基因敲除菌株E.coli ΔdctA苹果酸摄取功能,首先在以0.2%葡萄糖或0.2%苹果酸作为唯一碳源M9固体平板测试E.coli K12和E.coli ΔdctA生长。结果发现,E.coli K12在以上两种碳源M9固体平板均正常生长,相比之下,E.coli ΔdctA无法在以0.2%苹果酸作为唯一碳源M9固体平板生长(见图5A)。进一步测试E.coli ΔdctA转化子生长情况。结果发现,E.coli ΔdctA/pEasy T3-dctA在以上两种碳源M9固体平板均正常生长,相比之下,E.coli ΔdctA/pEasy T3(空载体作为负对照)无法在以0.2%苹果酸作为唯一碳源M9固体平板生长(见图5B)。在此基础上,绘制野生型E.coli K12和E.coli ΔdctA在以0.2%苹果酸作为唯一碳源M9液体培养基中生长曲线。结果发现,24 h内,E.coli K12生长达到稳定期,基因敲除菌株E.coli ΔdctA无法生长(见图5C)。因此,24 h为受试菌株培养时间。采用同样方法,测试24h内E.coliK12(正对照)、E.coliΔdctA(菌株作为负对照)、E.coli ΔdctA/pEasy T3(空载体作为负对照)和E.coli ΔdctA/pEasy T3-dctA在以0.2%苹果酸作为唯一碳源M9液体培养基中生长情况。结果发现,在以0.2%苹果酸作为唯一碳源M9液体培养dctA均正常生长,E.coli ΔdctA(菌株作为负对照)和E.coli ΔdctA/pEasy T3(空载体作为负对照)均无法生长(见图5D)。以上结果表明,E.coli ΔdctA丧失苹果酸摄取功能,即基因敲除菌株E.coli ΔdctA构建成功。四碳-二羧酸摄取蛋白DctA具有摄取苹果酸功能。
3 讨论与结论
在无氧条件下,大肠杆菌(E.coli)对四碳-二羧酸和L-天冬氨酸摄取、交换和外排由三个独立二羧酸摄取(Dcu)系统介导:DcuA,DcuB和DcuC[15-16]。但在有氧条件下,大肠杆菌(E.coli)摄取四碳-二羧酸由DctA介导[17]。DctA由电化学质子梯度提供驱动力,其活性受琥珀酸盐诱导及分解代谢物抑制[17]。DctA能否摄取四碳-2-羟基羧酸(如苹果酸)国内仍未见报道。本研究利用λ Red同源重组系统对野生型大肠杆菌(E.coli)K12 dctA作基因敲除,成功构建突变株E.coli ΔdctA。在含有以0.2%苹果酸为唯一碳源M9培养基中,通过功能互补测试证明E.coli ΔdctA丧失摄取苹果酸功能,四碳-二羧酸摄取蛋白DctA具有摄取苹果酸功能。
λ Red重组系统可实现基因靶向敲除,由gam,bet和exo三个基因编码。gam基因编码Gam蛋白,抑制大肠杆菌RecBCD蛋白复合体核酸外切酶活性,防止线性DNA片段被降解[18]。bet基因编码Beta蛋白是一个DNA单链结合蛋白,可与DNA单链结合促进互补链退火形成新双链分子,是一种重组蛋白[19]。exo基因编码Exo蛋白[20],其活性形式为一个环状三聚体,使环状相连三个单体分子内部形成通道,通道一端可容纳双链DNA分子,另一端仅可容纳单链DNA。Beta蛋白和Exo蛋白共同作用下完成线性DNA重组,最终实现基因敲除。本研究通过λ Red重组系统成功构建大肠杆菌K12 dctA基因敲除突变株E.coli ΔdctA,证明该方法敲除大肠杆菌基因方面应用可靠性较高,基因敲除突变株E.coli ΔdctA构建可为其他菌株来源2-HCT转运蛋白基因克隆与功能鉴定奠定基础。
[1] Niedringhaus T P,Milanova D,Kerby M B,et al.Landscape of next-generation sequencing technologies[J].Analytical Chemistry,2011,83(12):4327-4341.
[2] Shendure J,Ji H.Next-generation DNA sequencing[J].Nature Biotechnology,2008,26:1135-1145.
[3] 张森,李辉,顾志刚.功能基因组学研究的有力工具-比较基因组学[J].东北农业大学学报,2005,36(5):664-668.
[4] Nozaki K,Inaba K,Kuroda T,et al.Cloning and sequencing of the gene for Na+/H+Antiporter of Vibrio parahaemolyticus[J].Biochemical and Biophysical Research Communications,1996,222(3):774-779.
[5] Pinner E,Kotler Y,Padan E,et al.Physiological role of nhaB,a specific Na+/H+antiporter in Escherichia coli[J].Journal of Biological Chemistry,1993,268(3):1729-1734.
[6] 王爽,杨谦,孙磊,等.盐碱土中可培养中度嗜盐菌的研究[J].东北农业大学学报,2010,41(8):37-42.
[7] Jiang J,Pan Y,Meng L,et al.Halomonas zhaodongensis sp.nov.,a slightly halophilic bacterium isolated from saline-alkaline soils in Zhaodong,China[J].Antonie Van Leeuwenhoek,2013,104(5):685-694.
[8] Wang K,Zhang L,Yang Y,et al.Halobacillus andaensis sp.nov.,a moderately halophilic bacterium isolated from saline and alkaline soil[J].International Journal of Systematic and Evolutionary Microbiology,2015,65(6):1908-1914.
[9] Meng L,Meng F,Zhang R,et al.Characterization of a novel twocomponent Na+(Li+,K+)/H+antiporter from Halomonas zhaodongensis[J].Scientific Reports,2017,7(1):4221.
[10]Dong P,Wang L,Song N,et al.A UPF0118 family protein with uncharacterized function from the moderate halophile Halobacillus andaensis represents a novel class of Na+(Li+)/H+antiporter[J].Scientific Reports,2017,7:45936.
[11]Davies S J,Golby P,Omrani D,et al.Inactivation and regulation of the aerobic C4-dicarboxylate transport(dctA)gene of Escherichia coli[J].Journal of Bacteriology,1999,181(18):5624-5635.
[12]Sobczak I,Lolkema J S.The 2-hydroxycarboxylate transporter family:Physiology,structure,and mechanism[J].Microbiology and Molecular Biology Reviews,2005,69(4):665-695.
[13]Datsenko K A,Wanner B L.One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products[J].Proceedings of the National Academy of Sciences of the United States of America,2000,97(12):6640.
[14]彭玮,王聃,刘思国,等.大肠杆菌“菌影”的制备[J].东北农业大学学报,2009,40(6):85-88.
[15]Engel P,Krämer R,Unden G.Anaerobic fumarate transport in Escherichia coli by an fnr-dependent dicarboxylate uptake system which is different from aerobic dicarboxylate uptake[J].Journal of Bacteriology,1992,174(17):5533-5539.
[16]Golby P,Davies S,Kelly D J,et al.Identification and characterization of a two-component sensor-kinase and responseregulator system(DcuS-DcuR)controlling gene expression in response to C4-dicarboxylates in Escherichia coli[J].Journal of Bacteriology,1999,181(4):1238-1248.
[17]Kay W W,Kornberg H L.The uptake of C4-dicarboxylic acids by Escherichia coli[J].European Journal of Biochemistry,1971,18(2):274-281.
[18]Murphy K C.Lambda Gam protein inhibits the helicase and chistimulated recombination activities of Escherichia coli RecBCD enzyme[J].Journal of Bacteriology,1991,173(18):5808-5821.
[19]Li Z,Karakousis G,Chiu S K,et al.The beta protein of phage lambda promotes strand exchange[J].Journal of Molecular Biology,1998,276(4):733-744.
[20]Rhoades M,Meselson M.An endonuclease induced by bacteriophage lambda[J].Journal of Biological Chemistry,1973,248(2):521-527.
猜你喜欢
环境工程技术学报(2022年3期)2022-06-05 07:20:20
昆钢科技(2021年6期)2021-03-09 06:10:20
食品科学(2018年10期)2018-05-23 01:27:28
电源技术(2016年9期)2016-02-27 09:05:25
西南医科大学学报(2015年1期)2015-08-22 13:01:46
中国当代医药(2015年9期)2015-03-01 02:01:59
西南军医(2015年6期)2015-01-23 01:25:50
河南科技(2014年16期)2014-02-27 14:13:33
食品科学(2013年23期)2013-03-11 18:30:10
中国果业信息(2013年12期)2013-01-22 12:16:58
