
联吡啶衍生物芳基钌配合物的合成及其与DNA、蛋白质相互作用
2018-06-06 05:50王红艳董益利季徐顾秋予苏PeterSadler刘红科
无机化学学报 2018年6期
葛 超 王红艳 董益利 李 季徐 芸 顾秋予苏 志 钱 勇*, Peter J.Sadler 刘红科*,
(1南京师范大学化学与材料科学学院,南京 210023)(2Department of Chemistry,University of Warwick,Gibbet Hill Road,Coventry CV4 7AL,UK)
0 引 言
1965年,美国科学家Rosenberg首次发现顺铂能够抑制细胞生长并开展了抗肿瘤实验研究[1]。由于顺铂的抗药性及毒副作用,人们将研究扩展到了过渡金属配合物抗癌方面,已报道了很多新型具有低毒、高效抗癌活性的金属(如铂、钯、铑、铱、锇、钌等元素)配合物,其中钌配合物抗癌成为研究热点[2]。Sadler等发现双核芳基钌配合物对光敏感,具有结合光诱导细胞死亡和光活化荧光成像的潜力[3]。Dyson等设计了一系列的含膦配体的芳基钌配合物并研究芳烃结构变化对抗癌活性的影响[4]。
在螯合配体中,吡啶类化合物由于容易进行化学修饰得到系列衍生物,比其它氮杂环化合物更有优势,其与芳基钌配位一般有2种方式,N^N配位及C^N配位。值得注意的是,含有N,N-螯合配体的芳基钌配合物不仅在催化反应中表现优异,而且它们良好的抗癌活性也令人瞩目[5-6]。Sheldrick和同事已经报道了一系列具有N,N-螯合多吡啶基配体的铑和铱配合物[7],它们表现出良好的细胞毒性并通过配位、插入或双重结合的方式与DNA作用[8]。
抗癌配合物水解速率的大小对抗癌活性有重要影响,含有卤素离去基团的芳基钌配合物进入体内更容易在细胞质里发生水解而不是血液里,因为血液里Cl-浓度比较高抑制了配合物的水解 (大约100 mmol·L-1),而细胞质中的 Cl-浓度低(<25 mmol·L-1),促进水解,水解后带正电荷的水解产物可能进入细胞核与带负电荷的DNA作用造成DNA损伤,从而导致癌细胞凋亡[9]。DNA是抗癌化合物在细胞内的主要靶标之一,核酸和其他分子之间的相互作用是生命科学中的基本问题[10]。一些疾病的标志性物质和DNA靶向药物的相互作用机制涉及基因的复制、转录、突变和物种的相关变异等[11]。血清白蛋白是一种重要的生物分子,也是重要的血液蛋白质,其具有转运配体如脂肪酸、氨基酸、类固醇、金属离子和药物的能力[12-13]。药物-白蛋白在血流中的结合能力对药物的活体分布、游离浓度、代谢和排泄性质有重要影响,同时也影响化疗过程中的药物稳定性和毒性[14]。由于牛血清白蛋白与人血清白蛋白含90%的相同序列,性质差别小,其价格低,许多文献也用牛血清白蛋白进行实验,所以本文选择用牛血清白蛋白来研究配合物与蛋白的相互作用[15]。

Scheme 1 Synthetic route and general structures of complexes 1 and 2
研究导致配合物抗癌活性降低的因素也具有特别重要的意义。通过对配合物的水解、与靶标物质如DNA、蛋白等相互作用研究,可以让我们全面了解抗癌机理,进而在未来设计高效低毒配合物时避免不利因素[10]。本文合成了2种单核芳基钌联吡啶衍生物配合物[Ru(η6-p-cymene)(L1)Cl]Cl(1)和[Ru(η6-p-cymene)(L2)Cl]Cl(2)(L1=4,4′-dimethyl-2,2′-bipyridine,L2=4′-methyl-(2,2′-bipyridine)-4-carbaldehyde oxime),通过元素分析、ESI-MS和1H NMR进行了表征。用MTT法测试了配体及配合物对癌细胞及正常细胞的毒性,并用紫外光谱法和荧光光谱法研究了配合物与DNA、BSA的作用。在上述结果基础上探讨了影响配合物抗癌活性的因素,将为研究新药物、新靶点、新机制提供参考。
1 实验部分
1.1 试剂与仪器
实验所用溶剂均采用分析纯试剂,根据实验需要,试剂在使用前均按照《实验室化学品纯化手册(第五版)》[16]进行纯化。所有的反应均在氩气保护氛围下进行。 4,4′-dimethyl-2,2′-bipyridine(L1)购自晚晴化学品公司。根据文献方法[12-13]制备了配体4′-methyl-(2,2′-bipyridine)-4-carbaldehyde oxime(L2)。除特殊要求,所有测试均在室温下进行。1H NMR采用Bruker AVANCE 400 spectrometer(德国)测定,C、H、N元素分析使用元素分析仪(vario ELⅢ,贺力氏公司,德国)测定,ESI-MS使用LCQ FLEET电喷雾质谱仪(赛默飞世尔科技,美国)测定(Source Voltage 4.0 kV,Sheath Gas Flow Rate 8 arb,Capillary Voltage 70 V,Capillary Temp 275 K,流动相为甲醇,流速为200 pL·min-1,进样方式为直接进样,样品配成甲醇溶液),紫外吸收分光光谱仪为美国瓦里安公司生产的Cary 50仪器,荧光光谱是用PerkinElmer Ls-55荧光光谱仪测定。
1.2 配合物的合成与结构
1.2.1 [Ru(η6-p-cymene)(L1)Cl]Cl(1)的合成
芳基钌二聚体[RuCl2(η6-p-cymene)]2(122.4 mg,0.2 mmol)、配体(L1)(73.6 mg,0.4 mmol)溶于 10 mL甲醇中,在氩气氛围下,338 K回流12 h。反应结束后通过柱层析分离,CH2Cl2/MeOH(15∶1,V/V)为洗脱剂,产量为85.2 mg,产率为43.5%。1H NMR(CDCl3,400 MHz):9.52(2H,d,pyridine),8.03(2H,s,pyridine),7.54(2H,d,pyridine),6.17(2H,d,p-cymene),6.02(2H,d,p-cymene),2.70(1H,dt,CH),2.58(6H,s,CH3),2.27(3H,s,CH3),1.06(6H,d,CH3)。ESI-MS:[1-Cl-]+m/z的理论值为454.98,实验值为455.25。元素分析(%):C22H26N2RuCl2的理论值为 C 53.88,H 5.34,N 5.71;实测值为 C 53.28,H 5.28,N 5.86。
1.2.2 [Ru(η6-p-cymene)(L2)Cl]Cl(2)的合成
将芳基钌二聚体[RuCl2(η6-p-cymene)]2(122.4 mg,0.2 mmol)、配体 L2(85.6 mg,0.4 mmol)溶于甲醇中,在氩气氛围下,338 K回流12 h。反应结束后通过柱层析分离,CH2Cl2/MeOH(10∶1,V/V)为洗脱剂,产量为83.7 mg,产率为40.3%。1H NMR(DMSO-d6,400 MHz):12.41(1H,s,OH),9.50(1H,t,pyridine),9.38(1H,dd,pyridine),8.70(1H,d,pyridine),8.53(1H,s,pyridine),8.35(1H,d,pyridine),7.89(1H,dd,pyridine),7.67(1H,t,CH),6.29~6.19(2H,m,p-cymene),6.02~5.91(2H,m,p-cymene),2.62~2.55(4H,m,CH,CH3),2.18(3H,d,CH3),1.01~0.86(6H,m,CH3)。 ESI-MS:[2-Cl-]+m/z的理论值为483.98,实验值为484.25。元素分析(%):C22H25N3ORuCl2的理论值为 C 50.87,H 4.85,N 8.09;实测值为 C 49.83,H 5.23,N 7.82。
1.3 配合物的水解
通过UV-Vis光谱测定配合物1、2的水解。将配合物溶于DMSO,浓度为2 mmol·L-1,加水稀释,得浓度为50 μmol·L-1的溶液 (溶剂体积比为VDMSO∶VH2O=5∶95)。 在 298 K 下,于波长 200~800 nm 范围用1 cm路径的石英比色皿测定,12 h内每隔15 min记录吸光度值。
1.4 配合物与CT-DNA的相互作用
将 CT-DNA配制在磷酸盐缓冲液(PBS,5 mmol·L-1,pH 7.0)中,储存在 269 K 温度下。 用 PBS适当稀释后,通过UV-Vis在波长为260 nm处用1 cm路径的石英比色皿测量吸光度值,确定CT-DNA的浓度(ε=6 600 L·mol-1·cm-1)。紫外吸光强度的比值(A260/A280)约为1.9,表明DNA溶液不含蛋白质。滴定实验在室温下进行,用双通道进行测试,以相应浓度的CT-DNA作为空白对照。将50 μmol· L-1配合物溶液置于1 cm路径的石英比色皿中保持浓度不变,加入等体积的0.5 μL CT-DNA储备溶液,CTDNA与配合物的浓度比例CT-DNA与配合物的浓度比例 r 依次为 0.1、0.2、0.3、0.4、0.5、0.6、0.7、0.8、0.9、1.0,CT-DNA 的 浓 度 分 别 为 5、10、15、20、25、30、35、40、45、50 μmol·L-1。CT-DNA 与配合物混合7 min以达到平衡,于200~800 nm范围记录吸光度值。
1.5 配合物与血清白蛋白的相互作用
称取2 mg BSA,用pH=7.4的PBS溶解,加缓冲液稀释,配制浓度为3 μmol·L-1的BSA溶液。将配合物溶于DMSO,使配合物的初始浓度为10 mmol·L-1,将配合物用pH=7.4的PBS稀释至1.5 mmol·L-1,滴定实验在室温下进行,将 3 μmol·L-1的BSA溶液置于1 cm路径的石英比色皿中,加入等体积的0.5 μL配合物溶液,配合物与BSA的比例r依次为 0、0.5、1.0、1.5、2.0、2.5、3.0、3.5,配合物的浓度 分 别 为 0、1.5、3.0、4.5、6.0、7.5、9.0、10.5 μmol·L-1。混合10 min达到平衡后,于样品的激发波长285 nm处测定荧光强度。
1.6 细胞毒性研究
实验选取了3种细胞系:人卵巢癌细胞(A2780),肺癌细胞(A549)和正常肝脏细胞(L02),来源于中山大学(中国,广州)动物实验中心。细胞在含10%的血清培养基RPMI1640(Gibco BRL)中37℃培养。配合物对癌细胞体外抗增殖活性试验方法为:取对数生长期的细胞,调整细胞悬液浓度为5×104CFU·mL-1,每孔100 μL细胞悬液接种于 96孔细胞培养板中,预培养24 h。分别设空白对照组、细胞对照组以及 4 个浓度(12.5、25、50、100 μmol·L-1)配合物药物组,孵育48 h。然后向每个孔中加入20 μL噻唑蓝(MTT,5 mg·mL-1,以 PBS 溶解),37 ℃培养 4 h。小心地除去培养基,向每个孔中加入150 μL DMSO以溶解甲瓒晶体,避光放置30 min。使用酶标仪(Infinite M200,Tecan,Mannedorf,瑞士)测定 590 nm处的吸光度。计算配合物对癌细胞增殖(48 h)的半数抑制浓度(IC50)[17-18]。
2 结果与讨论
2.1 细胞毒性研究
采用MTT法测定了配体L1和L2及配合物1、2对肺癌细胞(A549)、人卵巢癌细胞(A2780)和正常肝脏细胞(L02)细胞毒性(IC50)。由配合物IC50结果(表1)可以看出,配体L1和L2对肺癌细胞(A549)有较强的毒性,其中 L1 的细胞毒性(11.83 μmol·L-1)优于临床使用的抗癌药物顺铂(13.84 μmol·L-1),但他们对正常细胞毒性很小(>100 μmol·L-1)。配合物 1 和 2对癌细胞A2780、A549和正常细胞L02基本没有毒性,远远低于顺铂的毒性。从上述结果可以看出,与对-伞花烃钌配位后,配合物1、2对癌细胞及正常肝脏细胞的毒性急剧降低(~200 μmol·L-1)。 为了探究造成配合物抗癌活性降低的原因,我们研究了配合物的水解及其与蛋白、DNA的相互作用。

表1 配体L1、L2及配合物1、2和顺铂的细胞毒性(IC50)Table 1 Cell toxicity of L1,L2 and complexes 1,2 and cisplatin (IC50)μmol·L-1
2.2 配合物的水解
配合物的水解化学可以调控它们的药理学性质,包括细胞摄取、分布、与DNA结合、体内代谢和毒性等[19-20]。通过紫外吸收光谱研究了1、2的水解(图1)。配合物1在250 nm处有一肩峰,264 nm处有较强的吸收峰可归属为混合LLCT/MLCT(配体ππ*/中心金属d轨道上的电子向配体π*轨道的跃迁)跃迁,在304和314 nm处的较强吸收峰归属为LLCT跃迁;配合物2在250和280 nm处的较强吸收峰可归属为混合LLCT/MLCT跃迁,320 nm处较弱的吸收峰可归属为LLCT跃迁[21]。2个配合物的水解都是离去基团Cl-与H2O发生交换,有新物质生成,产物浓度的升高会导致吸收峰增强,而配合物浓度的降低则导致吸收峰减弱。配合物1水解时的吸收峰强度随着时间变化很小,最大吸收峰处的吸光度略微增加(从0.551增加到0.557),说明1在水溶液中比较稳定,。配合物2水解时的吸收峰强度随时间变化比较明显,280 nm吸收峰处的吸光度有较大增加(从0.716升至0.759)。上述结果说明2个配合物水解反应不同,且配合物1水解速率比2慢。根据吸光度的变化,按准一级反应动力学[22]计算出配合物1和2的水解速率常数分别为0.383和1.458 h-1,半衰期分别为 1.81 和 0.47 h(表 2),与文献报道[22]的水解方式一样。
配合物1、2的水解反应的速率不同可能与其联吡啶螯合配体有关,配体L1和L2的取代基分别为-CH3和-CHNOH,-CHNOH为亲水基团,能增加配合物2的溶解度。

表2 配合物1和2的水解常数Table 2 Hydrolysis constant of complexes 1,2
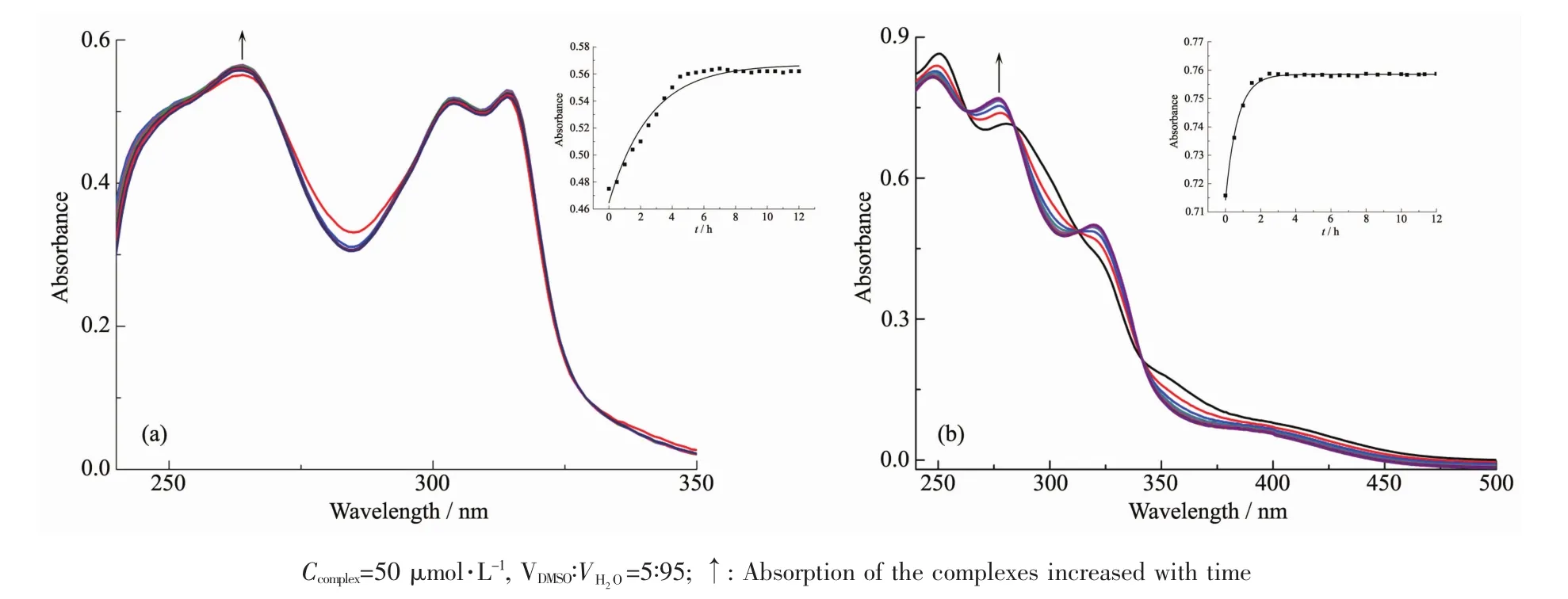
图1 配合物1(a)和2(b)在DMSO溶液中的紫外吸收光谱图Fig.1 Absorption spectra of complexes 1(a)and 2(b)in the DMSO solution
2.3 配合物与CT-DNA的相互作用
芳基钌配合物在细胞中的可能靶点是DNA,研究DNA和金属配合物之间的相互作用在抗癌机制中至关重要[23-24]。当配合物与DNA发生作用时,紫外光谱吸收峰会表现出减色现象,减色越明显表明配合物与DNA相互作用越强[25]。通过紫外光谱法研究了配合物1、2与DNA相互作用。在200~600 nm范围测定配合物1、2与CT-DNA作用的吸收光谱(图2)。 结果发现 1在250、263、304及 314 nm处有吸收,最强吸收峰出现在250和263 nm。配合物2在250、280及314 nm处有吸收,最强吸收峰出现在250和280 nm。1和2的紫外吸收强度随着DNA浓度的增加出现减色效应。配合物1和2的△A分别为0.05和0.1,减色百分比分别为8.3%和12.3%。2的减色效果强于1,出现这种差异的原因可能是联吡啶的取代基不一样[26]。配合物通过嵌入作用与DNA结合后与碱基对发生π电子堆积,配体的π*空轨道与DNA碱基对的π轨道发生偶合导致能级下降,偶合后的π*轨道部分填充电子,使其π-π*跃迁几率减小,从而产生减色效应[27]。上述结果表明配合物可能通过嵌入作用与双链CT-DNA结合。Schatzschneider等发现钌配合物紫外吸收峰波长的改变与联吡啶配体上的取代基有关,取代基会影响配体的π-π*的跃迁,取代基为吸电子基团时会使π-π*跃迁更容易[25]。配合物1和2中联吡啶配体上的取代基分别为-CH3和-CHNOH,-CHNOH为吸电子基,所以2的减色效果明显。
在紫外滴定实验基础上,利用以下公式计算了配合物 1和 2与 DNA 的结合常数 Kb:CDNA/(εa-εf)=CDNA/(εb-εf)+1/Kb(εb-εf)其中 εa是表观吸收系数,根据朗博-比尔定律,应为A/(bC),εb是完全结合DNA的消光系数,εf是没有加DNA时配合物的消光系数[28]。计算得到配合物1、2的结合常数分别为7.8×103和 1.86×104L·mol-1,说明配合物 2 与 DNA 的结合能力较强。造成这种差别的原因可能是配体L2的取代基为-CHNOH,-CHNOH是亲水基团,能增加配合物的溶解度,水解速度快。配合物1、2与DNA结合常数与文献报道的结果在同一个数量级[29],也表明配合物与DNA以嵌入方式作用。
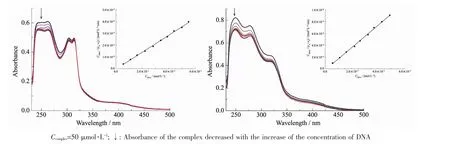
图2 配合物1(a)和2(b)加入DNA前后的紫外吸收光谱图Fig.2 Absorption spectra of complex 1(a)and 2(b)in the absence and the presence of CT-DNA
2.4 配合物与蛋白的相互作用
金属配合物与血清白蛋白BSA结合的定性分析通常通过荧光光谱进行检测。BSA产生荧光的生色团是由蛋白质的2个氨基酸引起的,即色氨酸(Trp)和酪氨酸(Tyr)。蛋白质构象转换,底物结合或变性反应都有可能导致色氨酸的发射光谱发生变化[30]。BSA的固有荧光可以提供关于其结构和动力学的信息,并且常用于蛋白质折叠和相关反应的研究[31]。配合物与BSA滴定实验的荧光发射光谱如图3(a)、3(b)所示。配合物1、2加入到BSA溶液中导致其在348 nm处的荧光强度的显著降低,分别降低了44.9%和31.8%(图4),说明配合物与血清白蛋白有较强的相互作用,且配合物1与蛋白质的结合能力强于2。荧光强度的变化可以用Stern-Volmer方程[32]描述:I0/I=1+kqτ0CQ=1+KsvCQ;其中 I0和 I分别是只有BSA时和猝灭剂(配合物)、BSA共存时荧光团的荧光强度,kq是Stern-Volmer淬灭常数,Ksv是动态猝灭常数,τ0为荧光团的荧光寿命,其平均寿命大约为10-8s,CQ为猝灭剂的浓度。根据此方程计算得到了配合物1和2的Ksv和kq。2个配合物的 kq分别为(7.50±0.47)×1012、(5.00±0.43)×1012L·mol-1·s-1(表3),远大于各类猝灭剂对生物大分子最大扩散碰撞猝灭速率常数(2×1010L·mol-1·s-1)[32-33],说明动态碰撞不是引起BSA荧光猝灭的主要原因,而是由于配合物和 BSA形成了不发光的加合物,从而引起了BSA内源荧光的静态猝灭。配合物1和2的Ksv值分别为(7.50±0.47)×104、(5.00±0.43)×104L·mol-1(表3),也说明配合物能与蛋白质发生作用,且配合物1与蛋白质的作用强于2。
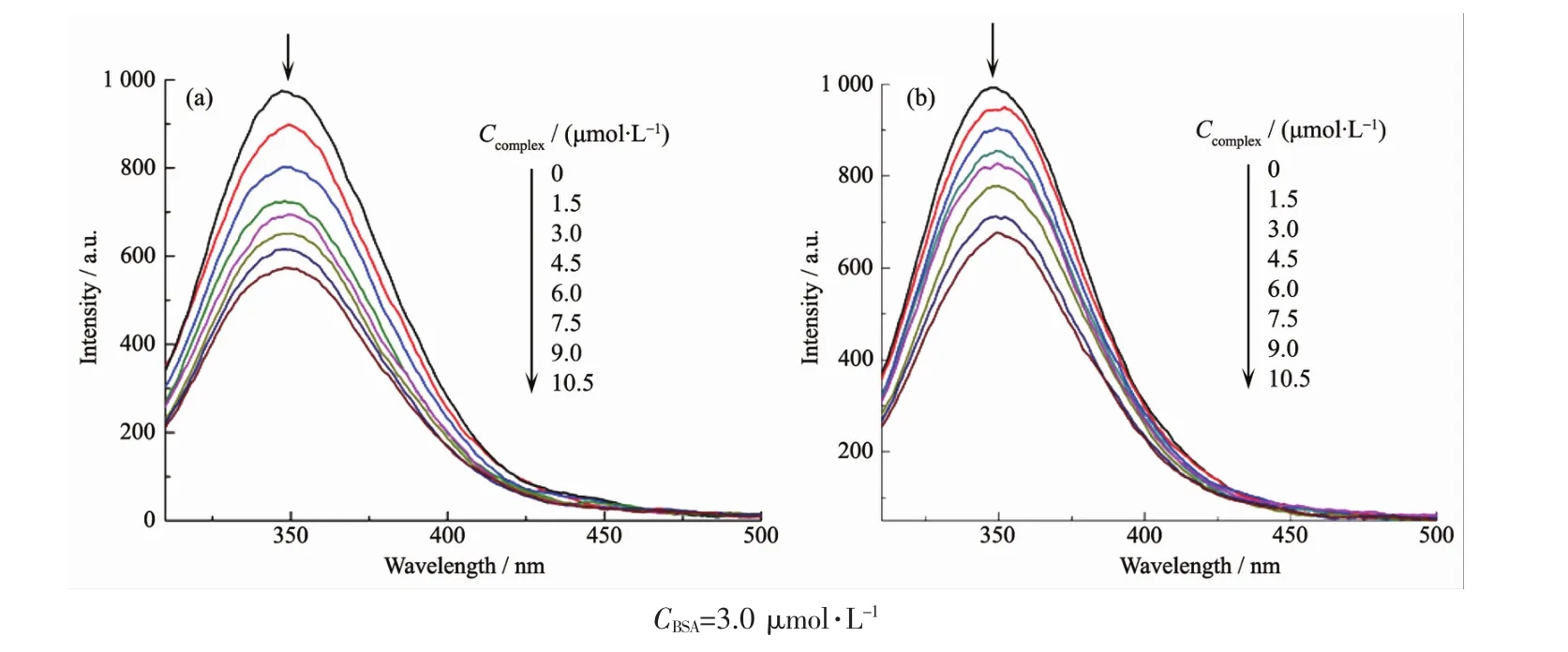
图3 BSA溶液中加入配合物1(a)和2(b)的荧光光谱Fig.3 Emission spectra of BSA in the absence and presence of increasing amount of complexes 1(a)and 2(b)

图4 BSA加入不同浓度的配合物1和2时的荧光强度变化Fig.4 Fluorescence intensity variation of BSA-bound complexes 1 and 2
假设蛋白质上有n个相同且独立的结合位点,根据 Scatchard 方程:Δ(I/I0)/CQ=nKA-KAΔ(I/I0)(CQ是猝灭剂即配合物的浓度),以 Δ(I/I0)/CQ对 Δ(I/I0)作图,根据截距和斜率分别可以求得配合物与BSA的结合常数KA和结合位点数n(表3),配合物 1、2的结合常数分别为(1.04±0.03)×105和(8.62±0.71)×104L·mol-1。这表明配合物与BSA有很强的相互作用,其中配合物1的作用较强。配合物与BSA之间的结合位点数约为1,即配合物与BSA分子存在1个结合位点。BSA主要有2个色氨酸残基发射内源荧光,一个是位于分子内部疏水腔的Trp-212;另一个是位于分子表面的Trp-134。由于Trp-134暴露在亲水环境会发生猝灭,所以BSA分子的内源荧光主要由处于疏水腔的Trp-212产生[34-35]。因此,可以推断引起BSA荧光猝灭的原因主要是配合物导致蛋白质二级结构发生变化,从而影响了Trp-212的疏水环境[36]。
血清白蛋白能够运输金属离子和金属配合物,它们之间相互作用可能增强或减弱配合物的生物特性和改变药物运输通路。如果配合物与血清白蛋白的结合常数KA太高,配合物就无法从血清白蛋白中释放进入靶细胞,导致细胞毒性降低[36]。配合物1和2与血清白蛋白的作用强于文献报道的结果[37],引起配合物的细胞毒性降低。其中配合物1的结合能力较强,造成这种差别的原因可能是配体L1的取代基为-CH3,-CH3是疏水基团,疏水基团可能进入BSA的疏水腔,与BSA分子以疏水作用方式结合,从而使色氨酸周围疏水性增加,引起蛋白分子的结合位点间相互作用[36]。

表3 配合物1和2对BSA的Stern-Volmer荧光猝灭数据Table 3 Stern-Volmer fluorescence quenching data of BSA in the complexes 1 and 2
3 结 论
利用双齿螯合结构的联吡啶类配体和芳基钌二聚体[RuCl2(η6-p-cymene)]2合成了单核配合物1和2,并探讨了影响其抗癌活性的因素。配合物2的水解反应速率比1快;1、2都能与DNA结合但配合物2的结合能力较强。配合物与蛋白的较强结合能力,可能是其细胞毒性不高的原因。配合物1的较慢水解速度有利于提高抗癌活性,其与蛋白结合能力较强,通过蛋白输送进入细胞,在细胞内水解进攻可能的靶标DNA;配合物2有可能在与蛋白结合之前已水解而与蛋白结合,没有到达靶标。上述结果可能是导致2个配合物抗癌活性略有不同的原因。因此,配合物的水解、与DNA和蛋白的作用可能是影响抗癌活性的主要原因。这些结果为未来设计新型高效的芳基钌抗癌药物将提供指导作用。
[1]Rosenberg B,Camp L V,Krigas T.Nature,1965,205:698-699
[2]Zhang C X,Lippard S J.Curr.Opin.Chem.Biol.,2003,7:481-489
[3]Magennis S W,Novakova O,Sadler P J,et al.Inorg.Chem.,2007,46:5059-5068
[4]Kilpin K J,Cammack S M,Dyson P J,et al.Dalton Trans.,2013,42:2008-2014
[5]Akitaka I,Nozomi K,Yoshio T,et al.Eur.J.Inorg.Chem.,2017,56:3794-3808
[6]Ashford D L,Glasson C R,Binstead R A,et al.Inorg.Chem.,2012,51:6428-6430
[7]Ornatsky O I,Lou X,Schfer S,et al.Anal.Chem.,2008,80:2539-2547
[8]Arockiam P B,Bruneau C,Dixneuf P H,et al.Chem.Rev.,2012,112:5879-5918
[9]Johnstone T C,Suntharalingam K,Lippard S J.Chem.Rev.,2016,116:3436-3486
[10]Muenzner K J,Kasparkova A,Schobert R,et al.J.Med.Chem.,2015,58:6283-6292
[11]Moggach S A,Prescimone A,Parsons S,et al.Inorg.Chem.,2007,46:4049-4059
[12]Furrer J,Süss-Fink G.Coord.Chem.Rev.,2016,309:36-50
[13]Kraatz H B,Keppler B K,Hartinger C G,et al.Inorg.Chem.,2010,49:7953-7963
[14]Mishra A,Mishra A,Ravikumar S,et al.Dalton Trans.,2014,43:6032-6040
[15]Singla R,Soni S,Padwad Y S,et al.Int.J.Biol.Macromol.,2017,104:748-757
[16]Armarego W L F,Chai C L L,Translated by LIN Ying-Jie(林英杰),LIU Wei(刘伟),WANG Hui-Ping(王会萍),et al.Purification of Laboratory Chemicals.5th Ed(实验室化学品纯化手册.5版).Beijing:Chemical Industry Press,2007.
[17]Rijt S H,Fu Y,Shnyder S D,et al.Metallomics,2014,6:1014-1022
[18]Li T,Aime S,Sadler P J,et al.Angew.Chem.Int.Ed.,2014,53:13225-13228
[19]Zhao M,Ji L N,Mao Z W,et al.Chem.Eur.J.,2013,19:12152-12160
[20]Kandioller W,Hartinger C G,Nazarov A A,et al.Organometallics,2009,28:4249-4251
[21]Vernooij R R,Kubeil C,Wood B R,et al.Inorg.Chem.,2017,56:5941-5952
[22]Habtemariam A,Moggach S A,Sadler P J,et al.Inorg.Chem.,2007,46:4049-4059
[23]Kubanik M,Kepplerb B K,Hartingeret C G,et al.Dalton Trans.,2016,45:13091-13103
[24]Heimann K,Dinh X T,Collins J G,et al.Mol.Biosyst.,2016,12:3032-3045
[25]Bischof C,Joshi T,Schatzschneider U,et al.Inorg.Chem.,2013,52:9297-9308
[26]Liu H K,Kong Y Q,Sadler P J,et al.Dalton Trans.,2016,45:18676
[27]Liu H K,Sadler P J.Acc.Chem.Res.,2011,44:349-359
[28]Chen L M,Peng F,Li G D,et al.J.Inorg.Biochem.,2016,156:64-74
[29]Wan Y,Zhang Y L,Li H L,et al.RSC Adv.,2014,4:35193
[30]Mohanraj M,Ayyannan G,Raja G,et al.Mater.Sci.Eng.C,2016,69:1297-1306
[31]Novakova O,Chen H,Sadler P J,et al.Chem.Biol.,2005,12:121-129
[32]Peng B H,Cui J G,Huang C H,et al.J.Org.Chem.,2015,783:10-16
[33]Peter T.Adv.Protein Chem.,1985,37:161-245
[34]GUO Qiong(郭 琼),LI Lian-Zhi(李 连 之),XU Tao(许 涛),et al.Acta Chim.Sinica(化学学报),2012,70:1617-1624
[35]KONG Ya-Qiong(孔亚琼),LI Ji(李季),LIU Hong-Ke(刘红科),et al.Scientia Sinica:Chimica(中国科学:化学),2017,47:277-283
[36]TanC,LiuJ,LiH,etal.J.Inorg.Biochem.,2008,102:347-358
[37]Gou Y,Zhang Y,Qi J X,et al.Oncotarget,2016,7:67004-67019
猜你喜欢
中南民族大学学报(自然科学版)(2022年6期)2022-11-02
分子催化(2022年1期)2022-11-02
小学生学习指导(低年级)(2021年6期)2021-07-19
中南民族大学学报(自然科学版)(2020年6期)2020-12-22
无机化学学报(2020年7期)2020-07-20
抗癌之窗(2020年1期)2020-05-21
特别健康(2018年9期)2018-09-26
无机化学学报(2018年8期)2018-08-01
中国塑料(2015年10期)2015-10-14
