
嵌合抗原受体T细胞联合免疫检查点抑制剂治疗恶性肿瘤的研究进展*
2018-06-01 07:00:44
中国肿瘤临床 2018年9期
近年来,肿瘤免疫治疗逐渐被证实具有较大的临床应用价值。经多项临床试验证实,嵌合抗原受体T细胞(chimeric antigen receptor T-cell,CAR-T)治疗明显提高了血液肿瘤的缓解率,使难治复发性急性淋巴细胞性白血病完全缓解率(complete response,CR)达90%[1],难治性大B细胞淋巴瘤(large B-cell lymphoma,B-DLCL)有效率达80%,18个月的总生存率(overall survival,OS)达52%[2]。在部分实体瘤治疗方面也表现出较好的治疗效果[3-4]。2017年8月美国食品药品监督管理局(FDA)批准Novartis公司第二代CAR-T细胞治疗用于治疗急性淋巴细胞白血病;2017年10月,FDA批准Kite制药的二代CAR-T细胞用于治疗包括弥漫性大B细胞淋巴瘤(diffuse large B cell lymphoma,DLBCL)在内的特定类型的 B-DL⁃CL[5-6]。此外,以抗PD-1为主的肿瘤免疫检查点疗法也在多项临床试验中取得较好的疗效[5-6],使复发难治性非霍奇金淋巴瘤(non-Hodgkin's lymphoma,NHL)反应率达65%~87%。上述免疫治疗策略在不同的恶性肿瘤中均产生较大且持久的抗肿瘤效应,以及广阔的临床研究前景。
1 CAR-T细胞治疗
1.1 CAR-T细胞治疗的发展
CAR-T细胞治疗目的是,通过对T淋巴细胞进行基因修饰使其表达特定的受体(CAR)以靶向结合特定的抗原,产生强大的免疫介导的抗肿瘤反应。与自然状态下的T细胞相比,CAR-T细胞识别肿瘤抗原绕过了抗原提呈阶段,因而不受MHC分子限制,避免了因肿瘤MHC分子下调而造成的逃逸[7]。
CAR结构主要有胞外抗体识别区和胞内信号转导区构成,两者通过跨膜区相连。目前,CAR-T细胞主要有四代。前三代的变化主要是胞内信号转导区域中所嵌入共刺激信号分子数量的不同:第一代CAR(不含共刺激信号分子)、第二代CAR(含有1个共刺激信号分子如CD28)和第三代CAR(含有2个或2个以上共刺激信号分子如CD28、CD137)。第4代CAR(TRUCK T细胞)在原有的抗体识别区和信号转导区基础上,增加一个或多个组成性或诱导性的表达组件,使CAR-T细胞表达特定的蛋白,如IL-12,以增强T细胞活性,并激活固有免疫细胞的抗肿瘤功能[8]。因此,第四代CAR-T细胞有调节肿瘤微环境的作用。另外,第四代CAR可以引入自杀基因,以便在发生如严重的细胞因子释放综合征等致命的不良反应时及时终止治疗[9]。现阶段应用于临床较多的CAR-T细胞治疗是第二代的CAR-T细胞。
1.2 CAR-T细胞治疗血液系统恶性肿瘤
CAR-T细胞疗法的临床试验多数集中于血液系统肿瘤,其中以CD19为靶点治疗B细胞肿瘤为主。CD19广泛表达于各个分化阶段的B细胞表面(从前体B细胞到记忆B细胞),并且促进B细胞的发育成熟,是理想的B细胞肿瘤的靶向抗原。杀伤CD19阳性细胞即使造成了正常B细胞的减少,也可应用免疫球蛋白进行替代治疗[7]。因此,靶向CD19 CAR-T细胞从治疗急性B淋巴细胞白血病(acute B lymphoblas⁃tic leukemia,B-ALL)扩展到多发性骨髓瘤和不同种类的NHL并且获得了较好的治疗效果,尤其对于复发难治性患者[10-11]。Neelapu等[11]报道了CAR-T细胞(自体抗CD19 CAR-T细胞)治疗复发难治性B细胞淋巴瘤(B cell lymphoma,BCL)的随访结果。该临床Ⅱ期试验共纳入111例BCL患者,经低剂量的环磷酰胺和氟达拉滨预处理后回输自体CAR-T细胞。中位随访时间为15.4个月,总反应率达82%,其中CR为54%,总生存期(overall survival,OS)达 18个月占52%[11]。除CD19外,还可作用于多种靶点杀伤血液系统肿瘤。选择CD22为杀伤靶点可使CD19弱表达或阴性的难治复发性B-ALL患者获得缓解[12]。另外,用于多发性骨髓瘤治疗的靶点有BCMA、CD138、CS1 等[13]。CD123、CLL-1(C-type lectin-like mole⁃cule-1)有望成为急性髓系白血病(acute myeloid leu⁃kemia,AML)治疗的新靶点[14-15]。
1.3 CAR-T细胞治疗实体瘤
CAR-T治疗实体瘤近年来也受到越来越多的关注。与血液系统肿瘤不同的是,CAR-T细胞在实体瘤发挥抗肿瘤作用中需要能够到达肿瘤内部并克服更强的免疫抑制作用[7]。目前,国内外CAR-T治疗实体瘤的研究日渐涌现,新的靶点层出不穷,研究前景广阔。Guo等[16]研究报道19例采用CAR-T-表皮生长因子受体(epidermal growth factor receptor,EGFR)细胞治疗EGFR阳性的晚期胆管癌,其中1例获得CR,10例获得疾病稳定(stable disease,SD),中位无进展生存期(median progression free survival,mPFS)为4(2.5~22.0)个月。Tchou等[17]报道了向转移性乳腺癌患者瘤体中注射c-Met-CAR-T细胞的治疗结果,在注射部位可检测到大片坏死的肿瘤细胞及巨噬细胞聚集等一系列的炎症反应。以IL13Rα2、HER-2/CMV和EGFRⅧ为靶点治疗恶性胶质瘤可改善病情[18]。NCT02442297、NCT02844062等多项CAR-T应用于复发难治恶性肿瘤治疗的临床试验正在开展中。虽然CAR-T细胞治疗可以提高复发难治恶性肿瘤的缓解率,但是对于淋巴瘤和实体瘤治疗效果尚有待提高,而且其高复发率导致治疗失败的问题亟待解决[19]。根据肿瘤免疫编辑的原理,免疫系统对肿瘤具有双重作用,不仅可以抑制其生长,也可以筛选出更适合在宿主体内存活的肿瘤细胞,从而发生逃逸[20]。通常,肿瘤细胞通过上调免疫检查点的表达,以促进免疫抑制微环境的形成,逃避免疫系统的清除作用[21]。因此,阻断T细胞的免疫检查点可能会优化CART细胞的治疗效果。
2 免疫检查点抑制治疗
免疫检查点是指免疫系统中存在的一些抑制性信号通路。正常情况下,免疫检查点的调节功能可以使机体免受自身免疫的攻击和炎症反应的损害。细胞恶变后可通过上调免疫检查点的表达,并与T细胞上相应的位点结合可明显抑制T细胞的杀伤活性,从而逃脱机体的免疫监视与杀伤,以维持自身的存活。免疫检查点疗法即通过阻断被癌细胞激活的免疫抑制通路,以提高T细胞抗肿瘤免疫应答的治疗方法。常见的免疫检查点有程序性死亡受体及其配体(programmed death-1,PD-1/programmed death-ligand 1,PD-L1)、细胞溶解性T淋巴细胞相关抗原-4(cytolytic T lymphocyte related antigen-4,CTLA-4)、B/T淋巴细胞衰减子(B/T lym⁃phocyte attenuator,BTLA)、淋巴细胞活化基因3(lym⁃phocyte activation gene 3,LAG-3),T细胞免疫球蛋白黏蛋白(T cells immunoglobulin domain and mucin domain protein-3,TIM-3)、腺苷 A2a受体(adenosine A2a re⁃ceptor,A2aR)等[22](图1)。其中PD-1/PD-L1和CTLA-4是目前研究较热的免疫检查点。
2.1 阻断PD-1/PD-L1的抗肿瘤作用
PD-1是表达在T细胞上的免疫检查点蛋白之一。PD-1与PD-L1的结合可以诱导磷酸化抑制T细胞受体下游的激活信号,从而限制T细胞增殖活性,降低其对肿瘤细胞的杀伤作用。另外,PD-1的免疫抑制作用不仅限于对T细胞,对其他淋巴细胞亚群的功能也有所影响,如促进调节性T细胞(Treg)的增殖和免疫抑制作用,抑制B细胞和自然杀伤细胞(NK细胞)的活性。因此,阻滞PD-1/PD-L1通路不仅可以增加T细胞数量,还可以通过增强细胞因子的分泌,减少Treg细胞和骨髓源性的抑制细胞(myeloid suppressor cells,MDSCs)来改变抑制性的肿瘤微环境[23]。Ribas等[24]研究发现黑色素瘤患者经派姆单抗(pembrolizumab),一种抗PD-1的单克隆抗体治疗后,肿瘤内部CD8+效应T细胞比例显著增多;而在治疗无反应的患者肿瘤中CD4+的效应T细胞比例增多。但在外周血中却未观察到相应的变化。Xu-Monette等[5]研究报道,抗PD-1治疗可使复发难治性霍奇金淋巴瘤(Hodgkin's lymphoma,HL)总反应率达到65%~87%,并且疾病可得到长期控制。但在复发难治性B细胞NHL中总反应率仅为35%,虽然可有一定程度的改善,但其治疗效果仍有待提高。
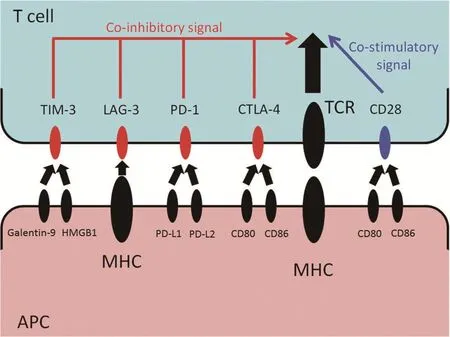
图1 免疫检查点对T细胞的影响[22]
2.2 阻断CTLA-4的抗肿瘤作用
CTLA-4是CD28-B7免疫球蛋白超家族成员。T细胞表达的CD28与APC表面的B7家族共刺激分子(B7-1和B7-2)的结合是激活T细胞重要的共刺激信号。当CTLA-4与B7家族的共刺激分子结合向T细胞提供抑制性信号,T细胞功能下降,并且应用抗CT⁃LA-4抗体可提高T细胞的抗肿瘤能力[25]。Ren等[26]研究报道,CTLA-4可降低抗CD20单抗治疗BCL的效果,阻滞CTLA-4与抗CD20单抗有协同抗肿瘤作用。Merchant等[27]研究报道17例伊匹单抗(ipilimum⁃ab)一种抗CTLA-4抗体,治疗儿童难治性恶性肿瘤的临床Ⅰ期试验,虽然未观察到肿瘤的消退,但有7例评效为SD,且观察到患儿血液循环中的T细胞有大幅增加。Sakamuri等[28]经过对多种难治性肿瘤的验证,发现抗CLTA-4抗体伊匹单抗与来那度胺有协同抗肿瘤作用。
2.3 其他的免疫检查点抑制剂
Zhou等[29]对59例经手术切除的肝细胞癌患者的癌组织研究发现,TIM-3、LAG-3、PD-1和CTLA-4在从癌组织中分离出的CD8+及CD4+T细胞表达均明显高于外周血及对照组;在肿瘤相关抗原(tumor associated antigen,TAA)特异性CD8+TIL的表达高于其他CD8+TIL。虽然,此类T细胞激活的水平高于未表达抑制性受体的T细胞,但其分泌的具有抗肿瘤作用的细胞因子和颗粒酶B却未增多,表明其活性受到一定程度的抑制。而且PD-1、TIM-3和LAG-3可促使TIL衰竭,应用相应的抗体可部分逆转衰竭信号的转导,明显地提高TIL的功能[29-30]。针对各种免疫检查点治疗的临床试验正在开展中(NCT03259867、NCT02694822和NCT03099109)。虽然已有越来越多的临床数据表明,抑制免疫检查点通路可以实现多种类型肿瘤的长期缓解[6],但由于肿瘤微环境复杂,单药治疗仅阻断其介导免疫抑制和免疫逃逸的效应,临床效果有限,肿瘤细胞仍可以通过其他途径实现免疫逃逸[31]。免疫检查点阻滞治疗应与其他治疗相结合,可以改善免疫抑制的肿瘤微环境并促进肿瘤反应性T细胞的扩增[32]。
3 免疫检查点抑制剂与CAR-T细胞的联合治疗
免疫检查点抑制与CAR-T细胞治疗均为增强T细胞抗肿瘤能力的治疗。如前所述,CAR-T细胞使T细胞绕过正常的活化途径,跨越了MHC分子的限制性,部分克服了肿瘤细胞的免疫逃逸,更易于活化进而杀伤肿瘤细胞;而免疫检查点在正常的免疫系统中相当于T细胞的“刹车系统”,在肿瘤微环境中,免疫检查点的过度表达限制了T细胞的杀伤力,因此阻滞免疫检查点信号通路相当于改造其“刹车系统”,使T细胞具有更强的抗肿瘤能力。上述两种治疗的结合可能会提高肿瘤治疗效果[33]。
3.1 PD-1与CAR-T细胞治疗的联合抗肿瘤作用
抑制T细胞功能或者耗竭T细胞均可造成CAR-T治疗的失败,PD-1/PD-L1部分介导了此效应[34]。在肿瘤特异性抗原刺激下,CAR-T细胞PD-1的表达会显著上调,造成其抗肿瘤免疫反应的削弱[34-35]。Gargett等[34]基于GD2-CAR-T细胞治疗转移性黑色素瘤的研究,发现经特异性抗原刺激后,在激活诱导性细胞死亡(ac⁃tivation-induced cell death,AICD)作用下,有活性的GD2-CAR-T细胞的比例下降,且下降程度与抗原表达水平呈正相关。虽然应用派姆单抗并未影响AICD的发生,但却可以使CAR-T细胞的活性恢复到抗原刺激之前的水平,同时促进了细胞因子的分泌[34]。Chong等[36]报道了1例难治性DLBCL经CD19 CAR-T治疗无反应加用派姆单抗后评效为疾病缓解(response disease,RD)的病例。并且观察到应用派姆单抗前,仅小部分CAR-T 19细胞保持增殖,大部分CAR-T19细胞未增殖或仅低水平增殖。加用派姆单抗后,CD19 CAR-T细胞的增殖活性有了大幅度提高。在2017年美国血液病协会(ASH)会议上,Chong等[37]报道,在所有经CAR-T治疗后首次应用派姆单抗的患者均有CAR-T细胞数量的增殖高峰,其中有应答者CAR-T细胞增殖有多个高峰,而无应答者仅在首次应用派姆单抗后表现出CAR-T细胞增殖速度的上升,其具体机制仍有待研究。抗PD-1抗体的应用虽然可以部分恢复CAR-T细胞的效应,明显减少肿瘤负荷,但在停止应用抗体后仍会有部分患者复发。因此,其效果较为短暂,需反复使用才能抑制肿瘤进展。为了实现持续性的不依赖于外源性抗体的PD-1阻滞,Li等[35]设计的CAR.αPD1-T细胞可分泌抗PD-1抗体,逆转了PD-1/PD-L1对T细胞功能的抑制效应,并提高了CAR-T细胞IFN-γ分泌的水平。Liu等[38]利用基因工程技术对CAR-T细胞进行了改造,使T细胞在胞外域同时表达PD-1及CAR结构。该研究当此PD-1与其配体PD-L1结合后可通过胞内的CD28传递激活信号,而非普通PD-1/PD-L1所传递的抑制性信号。相比派姆单抗阻滞PD-1,PD-1CD28使CAR-T细胞得以发挥更强的抗肿瘤效果。进一步的研究表明,派姆单抗虽然能阻滞PD-1/PD-L1且降低LAG+CD8TIL,但TIM3+/CECAM+肿瘤浸润性淋巴细胞(tumor infiltrating lym⁃phocyte,TIL)会有相应的增多;而用PD1 CD28修饰的CAR-T细胞则使LAG3表达及TIM3+/CECAM+共表达等下调,避免了因PD-1/PD-L1阻滞导致的其他免疫抑制因素的增强[38]。CAR-T联合PD-1抑制剂治疗恶性肿瘤的临床试验正在开展中(NCT03298828,NCT 02873390)。
Heczey等[39]研究则得出不同的结论,其课题组分别单独应用GD2-CAR-T细胞、GD2-CAR-T细胞加用环磷酰胺/氟达拉滨以及GD2-CAR-T细胞加用环磷酰胺/氟达拉滨和PD-1抑制剂治疗3组儿童神经母细胞瘤患者。相比于单独应用CAR-T治疗,环磷酰胺/氟达拉滨的应用使CAR-T细胞的增殖能力明显提高,但再加用PD-1抑制剂并未进一步提高其疗效。据此推测免疫检查点阻滞与CAR-T细胞治疗的协同作用,可能依赖于抑制PD-1的时间,或相比于儿童的恶性疾病(包括神经母细胞瘤),其需要更多的肿瘤抗原的刺激。因此,对于不同的患者群体及不同类型的肿瘤,其肿瘤微环境和对抗原的提呈能力均不同,CAR-T治疗与PD-1阻滞治疗联合应用与恶性肿瘤仍需进一步研究。
3.2 其他免疫检查点抑制剂与CAR-T细胞治疗的联合
除PD-1/PD-L1外,其他免疫检查点的表达也影响了CAR-T细胞抗肿瘤的效果。Condomines等[40]研究利用RNA干扰技术下调CTLA-4在CD19 CAR-T细胞上的表达,使其在体内扩增及抗肿瘤的能力均增强。有研究发现,LAG3、TIM3和PD-L1/PD-1的表达水平在CAR-T治疗无效的患者高于治疗有反应的患者[41]。Kenderian等[42]报道CAR-T细胞在与肿瘤细胞共培养1周后,TIM-3和PD-1表达均上调。阻滞TIM-3及PD-1后,CAR-T细胞产生细胞因子和Ki-67表达均增加。在移植白血病细胞的小鼠中联合应用CAR-T细胞及抗TIM-3抗体和抗PD-1抗体后,其缓解率显著高于单独应用CAR-T细胞者。Beavis等[43]研究发现,CAR-T细胞的激活导致A2AR的表达上调并抑制CAR-T细胞,这一现象可被A2AR拮抗剂或基因干扰所逆转,特别是与PD-1阻滞结合时。但这几种免疫检查点的阻滞与CAR-T治疗的结合尚在实验阶段,未见临床试验报道。
4 小结
CAR-T细胞治疗血液恶性肿瘤已经取得突破性进展,但其缓解率仍有待于进一步提高,复发率较高的问题亟待解决,而且CAR-T在实体瘤方面的应用仍需深入研究。免疫检查点抑制剂治疗恶性肿瘤已取得了较好的临床效果,但单药抗肿瘤效果较为有限,需与其他治疗相结合。最新的研究结果表明,阻滞免疫检查点可以在较大程度上改变CAR-T细胞所处的免疫抑制微环境,提高其增殖活性及抗肿瘤能力,改善多种复发难治恶性肿瘤预后。因此,CAR-T细胞联合免疫检查点抑制剂很有可能成为复发难治性肿瘤的重要治疗方法。
[1] Maude SL,Frey N,Shaw PA,et al.Chimeric antigen receptor T cells for sustained remissions in leukemia[J].NEngl J Med,2014,371(16):1507-1517.
[2] Neelapu SS,Locke FL,Bartlett NL,et al.Axicabtagene ciloleucel CAR T-Cell therapy in refractory large B-cell lymphoma[J].New Engl J Med,2017,377(26):2531-2544.
[3] Luo F,Qian J,Yang J,et al.Bifunctional alphaHER2/CD3 RNA-engineered CART-like human T cells specifically eliminate HER2(+)gastric cancer[J].Cell Res,2016,26(7):850-853.
[4] Brown CE,Alizadeh D,Starr R,et al.Regression of glioblastoma after chimeric antigen receptor T-cell therapy[J].N Engl J Med,2016,375(26):2561-2569.
[5] Xu-Monette ZY,Zhou J,Young KH.PD-1 expression and clinical PD-1 blockade in B-cell lymphomas[J].Blood,2018,131(1):68-83.
[6] Nghiem PT,Bhatia S,Lipson EJ,et al.PD-1 blockade with pembrolizumab in advanced merkel-cell carcinoma[J].N Engl J Med,2016,374(26):2542-2552.
[7] Lim WA,June CH.The principles of engineering immune cells to treat cancer[J].Cell,2017,168(4):724-740.
[8] Chmielewski M,Abken H.TRUCKs:the fourth generation of CARs[J].Exp Opin Biol Ther,2015,15(8):1145-1154.
[9] Tashiro H,Sauer T,Shum T,et al.Treatment of acute myeloid leukemia with T cells expressing chimeric antigen receptors directed to c-type lectin-like molecule 1[J].Mol Ther,2017,25(9):2202-2213.
[10]Garfall AL,Maus MV,Hwang WT,et al.Chimeric antigen receptor T cells against CD19 for multiple myeloma[J].N Engl J Med,2015,373(11):1040-1047.
[11]Neelapu SS,Locke FL,Bartlett NL,et al.Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma[J].N Engl J Med,2017,377(26):2531-2544.
[12]Fry TJ,Shah NN,Orentas RJ,et al.CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy[J].Nat Med,2017,(24):20.
[13]Mikkilineni L,Kochenderfer JN.Chimeric antigen receptor T-cell therapies for multiple myeloma[J].Blood,2017,130(24):2594.
[14]WangJ,ChenS,XiaoW,etal.CAR-TcellstargetingCLL-1asanapproach to treat acute myeloid leukemia[J].J Hematol Oncol,2018,11(1):7.
[15]Arcangeli S,Rotiroti MC,Bardelli M,et al.Balance of anti-CD123 chimeric antigen receptor binding affinity and density for the targeting of acute myeloid leukemia[J].Mol Ther,2017,25(8):1933-1945.
[16]Guo Y,Feng K,Liu Y,et al.PhaseⅠstudy of chimeric antigen receptor-modified T cells in patients with EGFR-positive advanced biliary tract cancers[J].Clin Cancer Res,2018,24(6):1277.
[17]Tchou J,Zhao Y,Levine BL,et al.Safety and efficacy of intratumoral injections of chimeric antigen receptor(CAR)T cells in metastatic breast cancer[J].Cancer Immunol Res,2017,5(12):1152-1161.
[18]Migliorini D,Dietrich PY,Stupp R,et al.CAR T-Cell therapies in glioblastoma:a first look[J].Clin Cancer Res,2018,24(3):535.
[19]Newick K,O'Brien S,Moon E,et al.CAR T cell therapy for solid tumors[J].Annu Rev Med,2017,(68):139-152.
[20]Schreiber RD,Old LJ,Smyth MJ.Cancer immunoediting:integrating immunity's roles in cancer suppression and promotion[J].Sci,2011,331(6024):1565-1570.
[21]Dunn GP,Old LJ,Schreiber RD.The Immunobiology of cancer immunosurveillance and immunoediting[J].Immunity,2004,21(2):137-148.
[22]Ok CY,Yong KH.Checkpoint inhibitors in hematological malignancies[J].J Hematol Oncol,2017,10(1):103.
[23]Zhu X,Lang J.Programmed death-1 pathway blockade produces a synergistic antitumor effect:combined application in ovarian cancer[J].J Gynecol Oncol,2017,28(5):e64.
[24]Ribas A,Shin DS,Zaretsky J,et al.PD-1 blockade expands intratumoral memory T cells[J].Cancer Immunol Res,2016,4(3):194-203.
[25]Leach DR,Krummel MF,Allison JP.Enhancement of antitumor immunity by CTLA-4 Blockade[J].Sci,1996,271(5256):1734-1736.
[26]Ren Z,Guo J,Liao J,et al.CTLA-4 limits anti-CD20-mediated tumor regression[J].Clin Cancer Res,2017,23(1):193-203.
[27]Merchant MS,Wright M,Baird K,et al.PhaseⅠclinical trial of ipilimumab in pediatric patients with advanced solid tumors[J].Clin Cancer Res,2016,22(6):1364-1370.
[28]Sakamuri D,Glitza IC,Betancourt Cuellar SL,et al.PhaseⅠdose-escalation study of anti-CTLA-4 antibody ipilimumab and lenalidomide in patients with advanced cancers[J].Mol Cancer Thera,2018,17(3):671.
[29]Zhou G,Sprengers D,Boor PPC,et al.Antibodies against immune checkpoint molecules restore functions of tumor-infiltrating T cells in hepatocellular carcinomas[J].Gastroenter,2017,153(4):1107-1119.
[30]Yang ZZ,Price-troska T,Novak AJ,et al.The exhausted intratumoral T cell population in B-cell non-hodgkin lymphoma is defined by LAG-3,PD-1 andtim-3 expression[J].Blood,2015,(126):2661-2661.
[31]Hanahan D,Weinberg RA.Hallmarks of cancer:the next generation[J].Cell,2011,144(5):646-674.
[32]Rosenblatt J,Avigan D.Targeting the PD-1/PD-L1 axis in multiple myeloma:a dream or a reality[J]?Blood,2017,129(3):275-279.
[33]Yeku OO,Purdon TJ,Koneru M,et al.Armored CAR-T cells enhance antitumor efficacy and overcome the tumor microenvironment[J].Sci Rep,2017,7(1):10541.
[34]Gargett T,Yu W,Dotti G,et al.GD2-specific CAR-T cells undergo potent activation and deletion following antigen encounter but can be protected from activation-induced cell death by PD-1 blockade[J].Mol Ther,2016,24(6):1135-1149.
[35]Li S,Siriwon N,Zhang X,et al.Enhanced cancer immunotherapy by chimeric antigen receptor-modified T cells engineered to secrete checkpoint inhibitors[J].Clin Cancer Res,2017,23(22):6982-6992.
[36]Chong EA,Melenhorst JJ,Lacey SF,et al.PD-1 blockade modulates chimeric antigen receptor(CAR)-modified T cells:refueling the CAR[J].Blood,2017,129(8):1039-1041.
[37]Chong EA,Melenhorst JJ,Svoboda J,et al.PhaseⅠ/Ⅱstudy of pembrolizumab for progressive diffuse large B cell lymphoma after anti-CD19 directed chimeric antigen receptor modified T cell therapy[J].Blood,2017,130(Suppl 1):4121-4121.
[38]Liu X,Ranganathan R,Jiang S,et al.A chimeric switch-receptor targeting PD1 augments the efficacy of second-generation CAR-T cells in advanced solid tumors[J].Cancer Res,2016,76(6):1578-1590.
[39]Heczey A,Louis CU,Savoldo B,et al.CAR-T cells administered in combination with lymphodepletion and PD-1 inhibition to patients with neuroblastoma[J].Mol Ther,2017,25(9):2214-2224.
[40]Condomines M,Arnason J,Benjamin R,et al.Tumor-targeted human T cells expressing CD28-based chimeric antigen receptors circumvent CTLA-4 inhibition[J].PLoS One,2015,10(6):e0130518.
[41]Schuster SJ,Svoboda J,Chong EA,et al.Chimeric antigen receptor T cells in refractory B-cell lymphomas[J].New Engl J Med,2017,377(26):2545-2554.
[42]Kenderian SS,Ruella M,Shestova O,et al.Identification of PD1 and TIM3 as checkpoints that limit chimeric antigen receptor T cell efficacy in leukemia[J].Blood,2015,126(23):852-852.
[43]Beavis PA,Henderson MA,Giuffrida L,et al.Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy[J].J Clin Invest,2017,127(3):929-941.
猜你喜欢
计算机系统应用(2022年4期)2022-05-10 08:41:10
中老年保健(2021年5期)2021-08-24 07:06:38
中老年保健(2021年6期)2021-08-24 06:53:48
中老年保健(2021年11期)2021-08-22 03:14:10
天津医科大学学报(2021年4期)2021-08-21 02:14:52
现代畜牧科技(2021年7期)2021-07-28 06:41:00
中华养生保健(2020年3期)2020-11-16 00:53:14
国际呼吸杂志(2019年4期)2019-03-12 01:08:18
兽医导刊(2019年1期)2019-02-21 01:13:54
中国药理学与毒理学杂志(2015年3期)2015-12-16 09:11:40
