
Autophagy inhibition: a new therapeutic target in spinal muscular atrophy
2018-05-31 09:02AntonioPiras,MarinaBoido
中国神经再生研究(英文版) 2018年5期
Spinal muscular atrophy (SMA) is a hereditary pediatric motor neuron(MN) disease: survival motor neuron 1 (SMN1) gene mutation deter‐mines MN degeneration and, consequently, muscle atrophy, breathing and swallowing difficulties, and, in the most severe cases, premature death. A second unaffected gene (SMN2) is present, but it can only pro‐duce a limited amount of functional protein, modulating the disease se‐verity and progression. SMN, ubiquitously expressed, is mainly involved in the assembly of small nuclear ribonucleoproteins and pre‐mRNA splicing requirements (Lunn and Wang, 2008). Its reduction determines selective MN loss: the type of cell death leading to neurodegeneration remains debated, since evidence of both increased apoptosis and dysreg‐ulated autophagy have been reported in SMA (Garcera et al., 2013).
Autophagy is an important intracellular process whereby cytoplasmic contents are delivered by double‐membrane vesicles, called autopha‐gosomes, to the lysosomes for degradation. This process is highly regulated by a series of proteins called autophagy‐related (ATG) proteins.Autophagic pathway controls degradation of cargo material such as damage mitochondria, damaged organelles, invading pathogens and aggregate‐prone proteins. However, increasing evidence underscore the relevance of autophagy in neuronal physiology and the involvement of autophagy‐lysosomal dysfunction in neurodegenerative disorders. In‐tracellular accumulation and aggregation of misfolded proteins, which are degraded by the autophagy‐lysosomal system, are reported in several pathological phenotypes of most late‐onset neurodegenerative disorders (reviewed in Menzies et al., 2017). On one hand, the activation of autophagy may be protective in some neurodegenerative diseases by the clearance of toxic proteins, on the other, its over‐activation or altered activity may perturb cell homeostasis and lead to cellular death.
Concerning SMA, somein vitroandin vivostudies have reported an increase of autophagic features in SMA MNs (Periyakaruppiah et al., 2016), suggesting that autophagy dysregulation can represent a new pathogenetic hypothesis in SMA and its pharmacological manipulation could in fluence the progression of the disease.
Involvement of autophagy in MN death in a mouse model of SMA:In order to investigate the role of autophagy in SMA, we used the SMNdel‐ta7 mouse, the most widely used and best‐characterized murine model(SMN2+/+; SMNdelta7+/+; SMN–/–), with an average lifespan of 13 days. In our work (Piras et al., 2017), we confirmed the increased expression of autophagic markers, Beclin 1 and microtubule‐associated protein 1 light chain 3‐II (LC3‐II), by western blot analysis and, furthermore, immu‐no fluorescence staining and electron microscopy studies demonstrated an increased number of autophagic vesicles (AVs) in the lumbar MNs of SMA pups. In addition, to evaluate the autophagic activity, we analyzed the level of p62/SQSTM1, which is selectively degraded by the autopha‐gosomes, observing that it was unchanged in the spinal cord of SMA compared to the control group. Altogether these results suggested that the autophagic flux was unaffected, thus hypothesizing that the accumulation of non‐functional AVs may lead the neurodegeneration in the spinal cord of SMA (Figure 1). Moreover, we observed an increased level of cleaved Caspase‐3 and reduced expression of Bcl2 (respectively, pro‐ and anti‐apoptotic markers), confirming an involvement of the apoptotic cell death in the SMNdelta7 mouse model as previously reported in human patients and otherin vivoSMA models (Figure 1).
However, so far no studies were conducted to address the direct role of autophagy in SMA and its pharmacological modulation in order to influence the disease progression. Several previous reports demonstrated that inhibition of autophagy by 3‐methyladenine (3‐MA) delayed neuronal loss in different mouse model of neurodegenerative disorders (e.g.,Alzheimer’s disease, Parkinson’s disease and Frontotemporal dementia)and ischemia (reviewed in Jaeger and Wyss‐Coray, 2009).
Thus, we evaluated the effect of the intracerebroventricular (ICV)administration of 3‐MA in our SMA model (Figure 2A) (Piras et al.,2017). We observed decreased levels of autophagic markers (Beclin 1 and LC3‐II) and reduction of AVs in the lumbar spinal cord after 3‐MA treatment. In accordance with these results, the protein levels of p62/SQSTM1 were increased. Altogether these results confirmed the effects of 3‐MA in the suppression of the autophagosome formation and au‐tophagic activity in the lumbar spinal cord of SMA pups. Interestingly,we found that 3‐MA administration reduced apoptotic cell death in the lumbar spinal cord, underlying the important relationship between auto‐phagy and apoptosis. Therefore, we performed stereological MN counts observing that ICV administration of 3‐MA improved MN survival in the lumbar spinal cord. Interestingly, inhibition of autophagy slightly,but significantly, increased the lifespan of SMA pups, and delayed the decrease in motor performance, in comparison to the control group treated with saline (Figure 2A).
Future prospective
Autophagy manipulation: therapeutic opportunities:Our observations strongly highlight the involvement of autophagy in SMA. We suggest for the first time that inhibition of this process by ICV administration of 3‐MA have a significant impact on disease progression, making autoph‐agy a novel and intriguing therapeutic target.
In agreement with previousin vivoandin vitrostudies (Garcera et al.,2013; Periyakaruppiah et al., 2016), we demonstrated an increase level of autophagosomes in the cytoplasm of MNs. In addition, since auto‐phagosome accumulation may affect the normal intracellular trafficking[ finally causing direct or indirect cytotoxicity (Figure 1)], we speculated that reducing autophagosome formation may improve MN survival in our mouse model of SMA.
Moreover, our results suggested that repeated administrations of 3‐MA were necessary to consistently delay the motor performance worsening in SMA pups compared to a single dose (Piras et al., 2017),thus suggesting that a prolonged therapy should be more indicated and necessary.
However, in this context, caution should be adopted in the application of 3‐MA: it inhibits autophagy acting temporally on class III phos‐phadylinositol 3‐kinase (PtdIns3K) and, on the contrary, it can equally promote autophagy especially after prolonged exposure by its action on the class I PtrIns3K. Thus, in a translational perspective, we suggest the use of tested molecules that directly modulate autophagy pathway with higher specificity,e.g. LYR294002 and Wortmannin (as discussed in Piras et al., 2017). As an alternative, genetic manipulation of autophagic genes could represent a potential strategy to modify the disease progres‐sion and pathogenesis, as already reported for other neurodegenerative disorders (reviewed in Loera‐Valencia et al., 2018).
Overall, our work strongly suggests that autophagy inhibition plays a protective role in SMA and, on the contrary, drugs that induce autopha‐gosome formation should be avoided for the treatment of SMA. However, additional studies are needed before clinical translation because, for example, manipulation of autophagy in the pathogenesis of amyotrophic lateral sclerosis (ALS, another MN disease) has been extensively investi‐gated in recent years showing controversial results (Ramesh and Pandey,2017). Indeed, preclinical studies on ALS models suggest that activation of autophagy represents a possible therapeutic target: several compounds promoting autophagy,e.g., rapamycin, lithium, and trehalose decrease aberrant protein aggregation in ALS. On the other hand, contradictory evidence has shown that rapamycin can worsen MN degeneration and decrease lifespan in a mouse model of ALS (Ramesh and Pandey, 2017).
In summary, all these results demonstrated that the autophagic process should be finely modulated in MN degenerative disorders, since it probably implicates a delicate balance between numerous molecules/mechanisms that, being detrimental or beneficial, retain opposing effects.
Genetic manipulation targeting SMN alternative splicing and combinatorial approaches:When several years ago the molecular genetic causes of SMA have been finally discovered, many drugs [including serine—arginine‐rich proteins, topoisomerase inhibitors, antisense oligonucle‐otides (ASOs) and histone deacetylase inhibitors] have been developed,mainly aimed at modifyingSMN2splicing: despite some encouraging preliminary results, many of them also showed disadvantages [as tox‐icity, short half‐life, insufficient specificity, inefficacy (Lunn and Wang,2008)], motivating the researchers to develop more effective, potent and specific molecules.
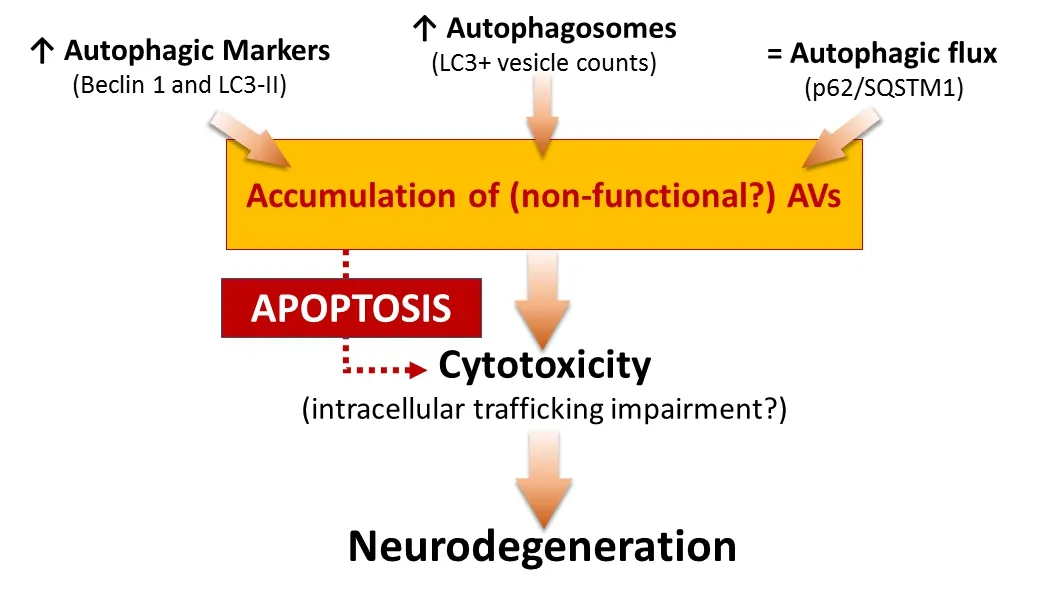
Figure 1 Summary and hypothesis of the role of autophagy in spinal muscular atrophy (SMA).
Looking at the most recent available therapeutic strategies, these are still aimed at increasing SMN expression, currently with very promising clinical results. Nusinersen, developed by Biogen and Ionis Pharma‐ceuticals, is the first‐ever treatment for SMA approved by the Food and Drug Administration: it is a potent ASO able to modulateSMN2splicing correction. Nusinersen can be delivered by lumbar puncture directly into the cerebrospinal fluid. The performed clinical trials provided evidence of its efficacy in significantly improving motor functions both in the most severe form of SMA (SMAI) and in milder patients(SMAII and III) (Mercuri et al., 2018), although the multiple and invasive intrathecal injections can induce adverse side effects. Similar results come from AVXS‐101, marketed by AveXis: it delivers theSMN1gene using non‐replicating self‐complementary AAV serotype 9. Compared to Nusinersen, AVXS‐101 has the great advantage to be delivered by intravenous injection. Its safety and efficacy have been proved in a clinical trial with SMAI patients, where the treatment assured the achievement of several motor milestones and lifespan extension (Tosolini and Sleigh,2017).
However, the above listed therapies are merely SMN‐dependent strategies, although it is now clear that further cellular mechanisms can affect the severity of SMA, in addition to SMN reduced expression. Since at now none therapeutic strategy aimed at restoring SMN is completely effective in arresting the disease progression, it is evident that combinatorial approaches are needed, keeping in mind the complexity of additional molecular pathways contributing to SMA pathogenesis. In such context autophagy can effectively represent a valid target, possibly in combination with gene therapy and ASO approaches.
Moreover, recent studies describe SMA as a multisystem disorder, in which in addition to the primary affected cells (i.e., spinal MNs) other non‐neuronal cells/organs are involved (Hamilton and Gillingwater,2013). Once again, the development of synergic treatments aimed i) to increase the SMN expression and ii) to prevent the disease progression using non‐SMN targets and SMN‐independent mechanisms, is clearly essential. Indeed, as concerns the autophagic activation, the process seems altered not only in the MNs as we observed, but also into SMA skeletal muscles (Oliván et al., 2016): therefore, the administration of an autophagic inhibitor systemically administered could further benefit the SMA prognosis.
Finally, despite the promising preliminary results, the ongoing clinical trials with Nusinersen still have some limitations mainly related to the strict eligibility criteria applied: since many milder SMA patients do not satisfy the requirements necessary for inclusion in gene therapy trials, alternative treatments should be rapidly provided for such individuals.
Therefore, in conclusion, for all these reasons, therapies targeting other molecular pathways (such as the autophagic process) are strongly required, alone or in combination with the emerging gene therapy and ASO approaches (Figure 2B), in order to assure the most effective treatment to all the SMA patients.
Antonio Piras*, Marina Boido
Neuroscience Institute Cavalieri Ottolenghi (NICO), Dept.
Neuroscience, University of Torino, Torino, Italy
*Correspondence to:Antonio Piras, Ph.D.,
antonio.piras@astrazeneca.com, current address is Innovative Medicines&Early Development - Respiratory, In flammation&Autoimmunity, AstraZeneca Mölndal, Sweden.
orcid:0000-0003-3052-1715 (Antonio Piras)
Accepted:2018-04-17
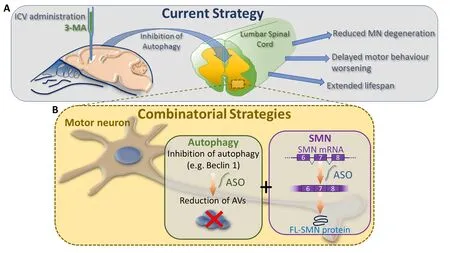
Figure 2 Current state and future therapeutic approaches.
doi:10.4103/1673-5374.232473
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer review reports:
Reviewer:Wei Wu, Indiana University, USA.
Comments to authors:In this manuscript, the authors introduced a new concept of inhibiting autophagy could be a potential therapeutic treatment for SMA. Authors briefly introduced the disease SMA and the role of autophagy in neuronal physiology and pathology. They reported their previous work on this topic as well: using 3-MA to inhibit autophagy in a widely accepted SMA model, effectively reduced apoptotic cell death in the spinal cord and increased the lifespan of SMA animals. In future prospective, they introduced recent studies on treatment of SMA including the discoveries from their group and other groups.
Garcera A, Bahi N, Periyakaruppiah A, Arumugam S, Soler RM (2013) Survival motor neuron protein reduction deregulates autophagy in spinal cord motoneurons in vitro. Cell Death Dis 4:e686.
Hamilton G, Gillingwater TH (2013) Spinal muscular atrophy: going beyond the mo‐tor neuron. Trends Mol Med 19:40‐50.
Jaeger PA, Wyss‐Coray T (2009) All‐you‐can‐eat: autophagy in neurodegeneration and neuroprotection. Mol Neurodegener 4:16.
Loera‐Valencia R, Piras A, Ismail MAM, Manchanda S, Eyjolfsdottir H, Saido TC,Johansson J, Eriksdotter M, Winblad B, Nilsson P (2018) Targeting Alzheimer’s disease with gene and cell therapies. J Intern Med doi: 10.1111/joim.12759.
Lunn MR, Wang CH (2008) Spinal muscular atrophy. Lancet 371:2120‐2133.
Menzies FM, Fleming A, Caricasole A, Bento CF, Andrews SP, Ashkenazi A, Füllgrabe J, Jackson A, Jimenez Sanchez M, Karabiyik C, Licitra F, Lopez Ramirez A, Pavel M,Puri C, Renna M, Ricketts T, Schlotawa L, Vicinanza M, Won H, Zhu Y, et al. (2017)Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic op‐portunities. Neuron 93:1015‐1034.
Mercuri E, Darras BT, Chiriboga CA, Day JW, Campbell C, Connolly AM, Iannaccone ST, Kirschner J, Kuntz NL, Saito K, Shieh PB, Tulinius M, Mazzone ES, Montes J, Bishop KM, Yang Q, Foster R, Gheuens S, Bennett CF, Farwell W, et al. (2018)Nusinersen versus sham control in later‐onset spinal muscular atrophy. N Engl J Med 378:625‐635.
Oliván S, Calvo AC, Rando A, Herrando‐Grabulosa M, Manzano R, Zaragoza P, Tiz‐zano EF, Aquilera J, Osta R (2016) Neuroprotective effect of non‐viral gene therapy treatment based on tetanus toxin C‐fragment in a severe mouse model of spinal muscular atrophy. Front Mol Neurosci 9:76.
Periyakaruppiah A, de la Fuente S, Arumugam S, Bahí N, Garcera A, Soler RM (2016)Autophagy modulators regulate survival motor neuron protein stability in moto‐neurons. Exp Neurol 283:287‐297.
Piras A, Schiaffino L, Boido M, Valsecchi V, Guglielmotto M, De Amicis E, Puyal J,Garcera A, Tamagno E, Soler RM, Vercelli A (2017) Inhibition of autophagy delays motoneuron degeneration and extends lifespan in a mouse model of spinal muscu‐lar atrophy. Cell Death Dis 8:3223.
Ramesh N, Pandey UB (2017) Autophagy dysregulation in ALS: when protein aggre‐gates get out of hand. Front Mol Neurosci 10:263.
Tosolini AP, Sleigh JN (2017) Motor neuron gene therapy: lessons from spinal muscular atrophy for amyotrophic lateral sclerosis. Front Mol Neurosci 10:405.
- 中国神经再生研究(英文版)的其它文章
- Novel function of the chemorepellent draxin as a regulator for hippocampal neurogenesis
- Weak phonation due to unknown injury of the corticobulbar tract in a patient with mild traumatic brain injury: a diffusion tensor tractography study
- Semaphorin 3A: from growth cone repellent to promoter of neuronal regeneration
- The role of undifferentiated adipose-derived stem cells in peripheral nerve repair
- Nerve conduction models in myelinated and unmyelinated nerves based on three-dimensional electrostatic interaction
- Fatigability during volitional walking in incomplete spinal cord injury: cardiorespiratory and motor performance considerations