
反相液相制备色谱法结合超临界流体制备色谱法分离纯化海风藤中的化合物
2018-05-28 07:36辛华夏彭子悦江大森梁鑫淼
色谱 2018年5期
辛华夏, 彭子悦, 江大森, 傅 青*, 金 郁, 梁鑫淼,2
(1. 华东理工大学药学院, 上海 200237; 2. 中国科学院分离分析化学重点实验室, 中国科学院大连化学物理研究所, 辽宁 大连 116023)
海风藤(piperkadsura)为胡椒科常绿攀缘藤本植物风藤的藤茎,已被广泛用于风湿、类风湿性关节炎和哮喘的治疗,是一种重要的藤类药物。海风藤中的化学成分十分丰富,主要包括木脂素、新木脂素、挥发油、生物碱、萜烯和多糖[1-5]。研究表明,海风藤中系列新木脂素类化合物具有抗神经炎活性[6],因此引起了人们对海风藤化学成分研究的兴趣。根据文献[7,8]报道,海风藤中化合物的分离基本采用传统的植物化学方法,如系统溶剂法、反复硅胶柱层析法等。在前期工作中,我们发展了基于反相液相色谱(reversed-phase liquid chromatography, RPLC)和超临界流体色谱(supercritical fluid chromatography, SFC)的分析方法,用于分离海风藤醇提物,SFC作为第二维色谱模式,具有快速、高效的特点[9]。为了更深入地了解海风藤的物质基础,有必要基于高效的分离技术发展相应的纯化方法,从而快速地在海风藤中获取更多化合物。
近年来,SFC的应用越来越广泛,其在手性拆分上具有巨大优势[10,11],张晶等[12]研究了涂覆型多糖手性固定相在SFC和HPLC上分离手性化合物的差异;李冬艳等[13]考察了改性剂种类和浓度对基于多糖固定相的SFC手性分离的影响。另外,SFC还在制药领域[14,15]和食品分析领域[16]表现出了应用潜力,也成为天然产物分析和制备的有力工具[17-19]。相比于高效液相色谱法(HPLC), SFC可以极大地缩短分析时间和节约溶剂用量[20,21]。由于超临界状态的CO2可以通过减压的方式快速去除,相比于制备HPLC,制备SFC的样品后处理更加简单、快速。此外,SFC与RPLC具有良好的分离正交性[22,23],两者可以组合成二维色谱,为复杂体系的分离提供更高的峰容量和更好的选择性。
本文的目的是发展RPLC和SFC的组合方法,用于海风藤中化合物的纯化,包括建立海风藤提取和前处理方案,考察和优化SFC条件,完成海风藤样品的RPLC制备和SFC制备以获得高纯度的化合物。
1 实验部分
1.1 仪器、试剂与材料
ACQUITY UPC2超高效超临界流体色谱仪(配备二元溶剂输送泵、自动进样装置、柱管理单元、二极管阵列检测器和背压调节器)、Waters SFC Prep 80超临界流体制备色谱(包括二元溶剂输送泵、自动进样装置、二极管阵列检测器、自动背压调节器、手动压力调节器和6个自动馏分收集器,数据采集和处理使用SuperChrom软件)、Alliance高效液相色谱(包括2695四元梯度泵、2489紫外可见检测器、自动进样和柱温箱,数据采集和处理用Waters Empower 3操作软件控制)、高效液相纯化系统(包括2545梯度泵、自动进样器、UV检测器、2767馏分收集系统和Mass Lynx V4.1色谱工作站)均购于美国Waters公司;BRUKER AVANCE Ⅲ-HD 600核磁仪(氢谱600 MHz、碳谱150 MHz)。
分析色谱柱Unitary C18(250 mm×4.6 mm, 5 μm)、XAmide(150 mm×4.6 mm, 5 μm,酰胺键合固定相)和制备色谱柱Unitary C18(250 mm×20 mm, 5 μm)、XAmide (250 mm×20 mm, 5 μm)均购于浙江华谱科技有限公司。
石油醚(分析纯,沸程:60~90 ℃)购于上海泰坦科技股份有限公司;甲醇(色谱纯)购于山东禹王实业有限公司;二氧化碳(食品级,纯度≥99.9%)购于上海振兴盖斯公司。实验用水由Milli-Q系统(美国Billerien公司)纯化。海风藤饮片样品购自河北安国中药材市场。
1.2 样品制备
将干燥的海风藤切碎。称取1.0 kg药材,加入10.0 L甲醇,回流提取1 h,过滤,然后在相同条件下重复提取1 h,合并两次提取液,浓缩至0.6 L,加入适量水使甲醇的体积分数为60%。叶绿素在该条件下沉淀析出,过滤除去。将滤液浓缩至干,加入10.0 g硅藻土及20.0 mL甲醇,搅拌均匀,风干。取干燥后的样品用500 mL石油醚提取1 h。
1.3 色谱条件
1.3.1RPLC制备条件
色谱柱:Unitary C18柱(250 mm×20 mm, 5 μm);流动相:(A)水和(B)甲醇;流速:19.0 mL/min;样品溶液质量浓度:250 g/L(甲醇溶解)。梯度洗脱程序:0~40 min, 55%B~70%B; 40~60 min, 70%B~100%B。进样体积:1.0 mL;检测波长:235 nm。按照色谱峰顺序依次收集保留时间10 min后的色谱峰,共计18个组分(F1~F18),每个组分减压蒸馏干燥后供SFC分离制备。
1.3.2SFC分析条件
XAmide色谱柱(150 mm×4.6 mm, 5 μm);流动相:(A)二氧化碳和(B)甲醇;柱温:30 ℃;流速:3.0 mL/min;梯度洗脱程序:0~10 min, 2%B~10%B;背压:13.8 MPa;进样体积:1.0 μL;检测波长:235 nm。
1.3.3SFC制备条件
XAmide色谱柱(250 mm×20 mm, 5 μm);柱温:30 ℃;流动相:(A)二氧化碳和(B)甲醇;流速:60 g/min;背压:15.0 MPa;进样体积:1.0 mL;检测波长:235 nm。F3、F8和F12组分的样品质量浓度分别为200、180和150 g/L(甲醇溶解)。梯度洗脱程序:F3, 0~12 min, 3%B~8%B; F8, 0~6 min, 5%B~8%B; F12, 0~12 min, 3%B~8%B。

图 2 石油醚层中海风藤在不同色谱柱上的色谱图Fig. 2 Chromatograms of the piper kadsura extracted by petroleum ether by the different chromatographic columns Modifier: methanol; backpressure: 13.8 MPa; column temperature: 30 ℃; detection wavelength: 235 nm. For columns BEH, BEH 2-EP and CSH FP (50 mm×2.1 mm, 1.7 μm), gradient elution program: 0-5 min, 1%-5% modifier in CO2; flow rate: 0.8 mL/min; injection volume: 0.2 μL. For column XAmide (150 mm×4.6 mm, 5 μm), gradient elution program: 0-5 min, 3%-8% modifier in CO2; flow rate: 3.0 mL/min; injection volume: 0.5 μL.
2 结果与讨论
2.1 样品前处理
海风藤的甲醇提取液呈墨绿色,其中含有大量叶绿素,本文采用醇提水沉的方法将其除去。实验考察了样品溶液中不同体积分数的甲醇对叶绿素去除效果的影响。结果表明,当甲醇的体积分数为60%时,叶绿素去除效果明显,样品溶液变为澄明的橙红色,液相色谱监测显示没有损失目标成分。为了进一步降低样品复杂性、富集目标成分,将上述样品浓缩后加入硅藻土进行分散,然后采用石油醚提取,使主要的目标成分集中在石油醚层。经以上前处理步骤,实验去除了样品中大量的叶绿素和极性相对较强的杂质,减少了在制备过程中对色谱柱的污染和对分离的影响,同时改善了样品的溶解性。
2.2 反相液相制备色谱
在甲醇和水的流动相体系下,石油醚层中海风藤在C18色谱柱上有很好的保留,物质的分离度和峰形都比较理想。该样品在甲醇中溶解度良好,可配制最大质量浓度为250 g/L的溶液。采用Unitary C18色谱柱(250 mm×20 mm, 5 μm)对样品进行分离,每次的进样量为1.0 mL,流速为19.0 mL/min。石油醚层中海风藤的反相制备色谱图见图1,显示样品在制备水平上分离良好。为了防止组分交叉,实验按照色谱峰顺序依次收集馏分,如图1所示,从10 min左右的色谱峰开始收集(该流速条件下检测延迟为2 s,收集时扣除延迟时间),共得到18个组分,依次编号为F1~F18。
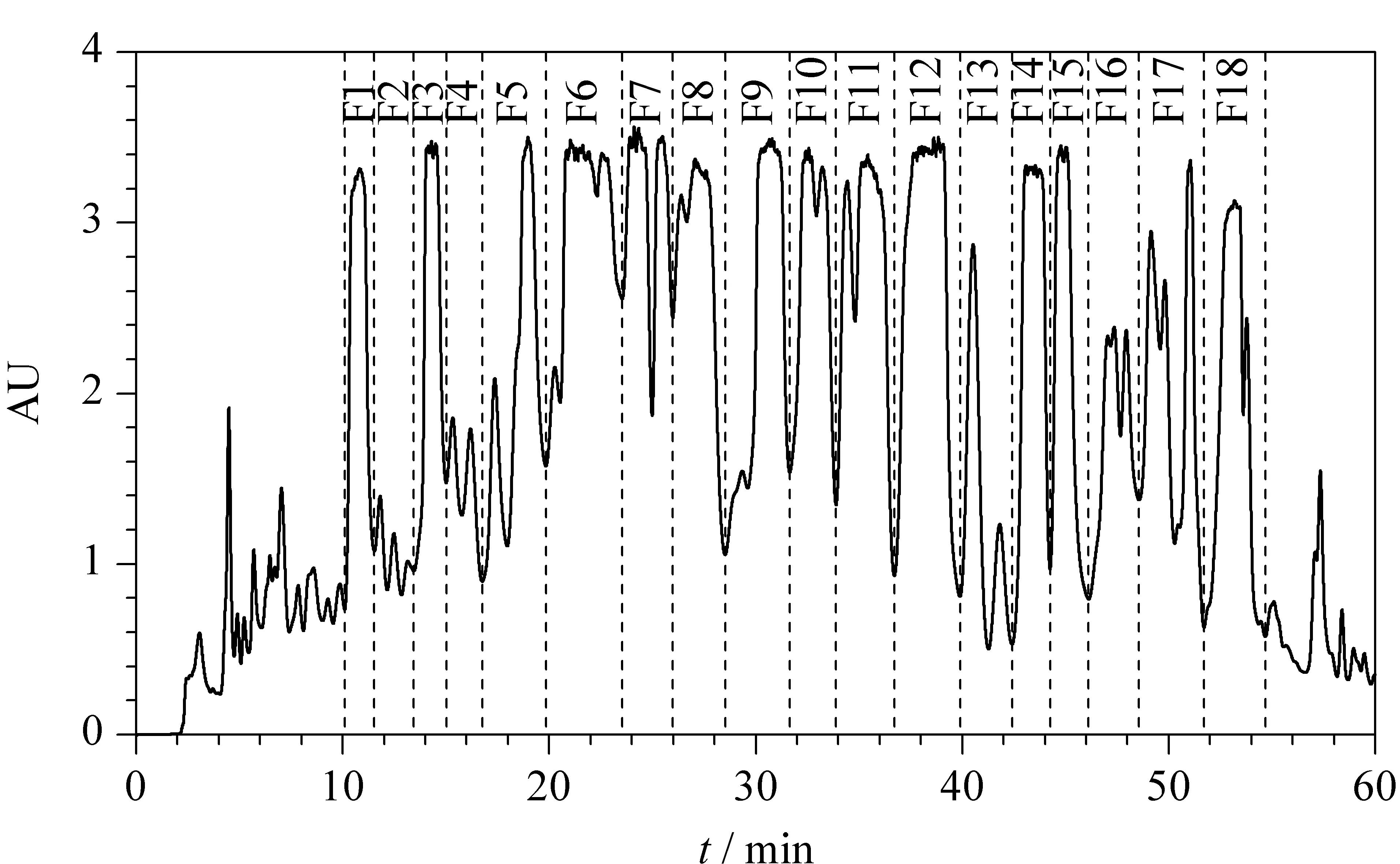
图 1 石油醚层中海风藤的反相制备色谱图Fig. 1 Preparation-RPLC (prep-RPLC) chromatogram of the piper kadsura extracted by petroleum ether Column: Unitary C18 (250 mm×20 mm, 5 μm); mobile phase: (A) water and (B) methanol; gradient elution program: 0-40 min, 55%B-70%B, 40-60 min, 70%B-100%B; injection volume; 1.0 mL (250 g/L methanol solution); flow rate: 19.0 mL/min; UV detection wavelength: 235 nm.
2.3 超临界流体色谱条件的优化
虽然经过了反相制备色谱的高效分离,但是所得到的组分仍很复杂,为了得到纯度较高的化合物,还需要进一步纯化。基于前期工作,本实验采用SFC进行第二次制备。以石油醚层中的海风藤为实验对象,首先对4种常见的色谱柱BEH(杂化硅胶,50 mm×2.1 mm, 1.7 μm)、BEH 2-EP(杂化硅胶表面键合2-乙基吡啶,50 mm×2.1 mm, 1.7 μm)、CSH-FP(杂化硅胶表面带电且键合氟苯基团,50 mm×2.1 mm, 1.7 μm)和XAmide(150 mm×4.6 mm, 5 μm)进行筛选(见图2)。在相同洗脱条件下,采用BEH 2-EP色谱柱时样品的保留时间最短,但分离选择性和采用BEH色谱柱时较接近;样品在CSH FP色谱柱上的保留和BEH色谱柱接近,分离效果比BEH和BEH 2-EP色谱柱好;相比于CSH FP色谱柱,XAmide色谱柱表现出了更好的保留和分离。在制备SFC中,采用较高比例的改性剂有利于样品溶解和提高制备仪器的稳定性,也有利于从分析到制备方法的转换[22]。采用XAmide色谱柱时改性剂甲醇的体积分数由1%~ 3%变为5%~8%。因此选择XAmide色谱柱用于该样品的SFC纯化分离。
除色谱柱的选择外,还需要考虑改性剂、柱温和背压对SFC分离效果的影响。和HPLC不同,在制备SFC中,样品不能采用初始流动相溶解,进样时可能会因为溶解度差异导致色谱峰变形,甚至在柱头析出,引起压力突然增高等问题。样品在甲醇中溶解度良好,因此本实验采用甲醇作为改性剂和样品溶解溶剂。根据文献[24,25]报道,适当降低柱温也有利于实验从分析到制备方法的转换,因此实验将柱温设定为30 ℃。压力是SFC的特征实验参数,在制备SFC上设置背压为15.0 MPa,在该背压条件下组分能得到较好分离,且制备过程稳定性较高。

图 3 组分F1~F18的三维SFC色谱图Fig. 3 Three-dimensional supercritical fluid chromatographic (SFC) chromatograms of the fractions F1-F18 Column: XAmide (150 mm×4.6 mm, 5 μm); modifier: methanol: gradient elution program: 0-10 min, 2%-10% modifier in CO2; flow rate: 3.0 mL/min; injection volume: 1.0 μL.
采用优化后的SFC条件对18个组分进行了分析(见图3)。如图3所示,经SFC分离后,得到了大量的色谱峰,除了响应强度较高的主峰外,响应强度较小的色谱峰也被有效富集和检测。结果说明SFC与RPLC高度正交,采用制备SFC可获得高纯度的化合物。
2.4 超临界流体制备色谱
实验选择了组成相对简单且含量较高的组分进行SFC制备。组分F12在反相模式下呈现一个主峰,主峰两侧有两个响应强度较小的色谱峰,面积归一化法显示主峰纯度为93.3%。在制备SFC色谱图中,除了主峰外,有多个响应强度较小的色谱峰被分离出来(见图4),说明SFC和RPLC具有良好的正交性,在反相模式下被主峰掩盖的一些小峰在SFC制备中被分离出来。SFC制备色谱图上主峰的峰宽为2 min,根据制备流速(60.0 g/min)和线性洗脱梯度,计算出每处理150 mg样品,收集的流分(甲醇)体积约为9.1 mL,和制备HPLC相比,溶剂消耗量大大降低。收集的流分中超临界二氧化碳通过挥发除去,甲醇则通过减压旋蒸除去。经检测,SFC制备色谱图中主峰的纯度提高至99.4%,经核磁鉴定并与文献[26]数据对比,确定该化合物为海风藤酮(kadsurenone)。
一些在反相模式下分离效果不理想的色谱峰在SFC上可以得到充分分离。在反相模式下,组分F8的保留时间为27 min,仍有肩峰的存在(见图4),通过优化洗脱条件以改善分离度较难。由制备SFC色谱图中可以看出,在5 min之内已快速完成了对该组分的分离,并得到4个分离度良好的色谱峰F8-1~F8-4。其中F8-1和F8-2根据峰面积积分,其色谱纯度均大于95.0%,经核磁鉴定和与文献[26]数据对比,确定为其分别为南藤素(wallichinine)和玉兰酯B(denudatin B)。而F8-3和F8-4的不对称峰形表明它们可能仍是混合物。
主峰和邻近小峰的分离是最为困难的,以组分F3为例,在反相模式下,组分F3呈现一个主峰,在主峰相邻的位置有一些未能分离的小峰。如果采用反相色谱进一步纯化,会损失较多的样品,另外,收集的主峰也难以达到较高的纯度。当采用制备SFC时,在12 min内可完成主峰和其附近小峰的分离,并且部分小峰之间的分离度和峰形也较好,F3-1、F3-3、F3-4和F3-6的色谱纯度均在95.0%以上。但峰F3-3因为含量较低,无法进行结构鉴定,峰F3-1、F3-4和F3-6经核磁鉴定和与文献[27-29]数据对比,确定为墙草碱(pellitorine)、2E-葵烯酸N-异丁基酰胺(2E-decenoic acidN-isobutylamide)和巴豆环氧素(futoxide)。分离得到的各个化合物的结构式见图5。
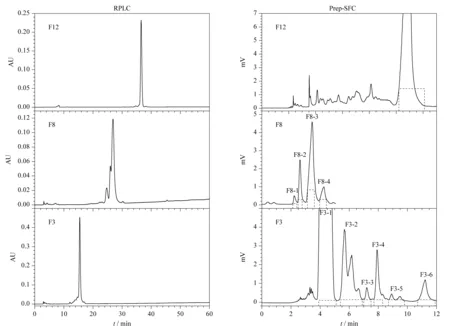
图 4 组分F12、F8和F3的RPLC分析色谱图和SFC制备色谱图Fig. 4 RPLC analytical and prep-SFC chromatograms of fraction F12, F8 and F3 RPLC conditions: column, Unitary C18 (250 mm×4.6 mm, 5 μm); mobile phase A, water; mobile phase B, methanol; gradient elution program, 0-40 min, 55%B to 70%B, 40-60 min, 70%B to 100%B; injection volume, 10.0 μL; flow rate, 1.0 mL/min; UV detection wavelength, 235 nm. Prep-SFC conditions: column, XAmide (250 mm×20 mm, 5 μm); modifier, methanol; flow rate, 60.0 g/min; back pressure, 15.0 MPa; column temperature, 30 ℃; UV detection wavelength, 235 nm. For F12, gradient elution program: 0-12 min, 3%-8% methanol; injection volume: 1.0 mL of 150 g/L. For F8, gradient elution program: 0-6 min, 5%-8% methanol; injection volume: 1.0 mL of 180 g/L. For F3, gradient elution program: 0-12 min, 3%-8% methanol; injection volume: 1.0 mL of 200 g/L.

图 5 海风藤中分离得到的化合物的结构式Fig. 5 Structural formulas of compounds separated from piper kadsura
3 结论
本文以石油醚提取层中海风藤的醇提物为研究对象,建立了样品前处理、RPLC制备和SFC制备的系统方法。海风藤样品在SFC模式中有很好的保留和分离,与RPLC进行组合后,利用两者不同的分离选择性可以实现主峰的纯化,难分离物质的分离,特别是微量成分的富集和制备。本文所建立的方法为深入研究海风藤化学成分提供可行性制备方案,同时拓展了SFC在天然产物分离领域的应用范围。
:
[1] Song J L, Yuan L, Liu Y J, et al. Journal of Hubei University of Chinese Medicine, 2007, 9(3): 70
宋敬丽, 袁林, 刘艳菊, 等. 湖北中医学院学报, 2007, 9(3): 70
[2] Ma Y, Han G Q, Liu Z J. Acta Pharmaceutica Sinica, 1993, 28(3): 207
马迎, 韩桂秋, 刘志坚. 药学学报, 1993, 28(3): 207
[3] Ma Y, Han G Q, Wang Y Y. Acta Pharmaceutica Sinica, 1993, 28(5): 370
马迎, 韩桂秋, 王银叶. 药学学报, 1993, 28(5): 370
[4] Lin L C, Shen C C, Shen Y C, et al. J Nat Prod, 2006, 69, 842
[5] Li X M, Wang D Y, Liu H Q. Chinese Traditional Paten Medicine, 2005, 27(10): 1199
李晓蒙, 王定勇, 刘慧琼. 中成药, 2005, 27(10): 1199
[6] Kim K H, Choi J W, Ha S K, et al. Bioorg Med Chem Lett, 2010, 20, 409
[7] Zhang S X, Liu X K, Zhang L, et al. Natural Product Research and Development, 1992, 4(3): 1
张顺祥, 刘星楷, 张莉, 等. 天然产物研究与开发, 1992, 4(3): 1
[8] Yuan L. [MS Dissertation]. Hubei: Hubei University of Chinese Medicine, 2008
袁林. [硕士学位论文]. 湖北: 湖北中医药大学, 2008
[9] Xin H X, Fu Q, Yuan Y, et al. J Supercrit Fluid, 2017, 127: 9
[10] Kalikova K, Slechtova T, Vozka J, et al. Anal Chim Acta, 2014, 821: 1
[11] Miller L. J Chromatogr A, 2012, 1250: 250
[12] Zhang J, Chen X D, Li L Q, et al. Chinese Journal of Chromatography, 2016, 34(3): 321
张晶, 陈晓东, 李丽群, 等. 色谱, 2016, 34(3): 321
[13] Li D Y, Wu X, Hao F L, et al. Chinese Journal of Chromatography, 2016, 34(1): 80
李冬艳, 吴锡, 郝芳丽, 等. 色谱, 2016, 34(1): 80
[14] Desfontaine V, Guillarme D, Francotte E, et al. J Pharm Biomed Anal, 2015, 113: 56
[15] Lemasson E, Bertin S, West C. J Sep Sci, 2016, 39(1): 212
[16] Bernal J L, Martin M T, Toribio L. J Chromatogr A, 2013, 1313: 24
[17] Hartmann A, Ganzera M. Planta Med, 2015, 81(17): 1570
[18] Li Z Y, Fu Q, Li K Y, et al. Chinese Journal of Chromatography, 2014, 32(5): 506
李振宇, 傅青, 李奎永, 等. 色谱, 2014, 32(5): 506
[19] Yuan Y, Xin H X, Peng Z Y, et al. Chinese Journal of Chromatography, 2017, 35(7): 683
袁云, 辛华夏, 彭子悦, 等. 色谱, 2017, 35(7): 683
[20] Miller L. J Chromatogr A, 2014, 1363: 323
[21] Miller L. J Chromatogr A, 2012, 1256: 261
[22] Sarrut M, Corgier A, Cretier G, et al. J Chromatogr A, 2015, 1402: 124
[23] Yang B C, Xin H X, Wang F E, et al. J Sep Sci, 2017, 40(16): 3231
[24] Tarafder A, Hill J F. J Chromatogr A, 2017, 1482: 65
[25] Tarafder A, Hudalla C, Iraneta P, et al. J Chromatogr A, 2014, 1362: 278
[26] Duan S T. [MS Dissertation]. Shanghai: Fudan University, 2009
段书涛. [硕士学位论文]. 上海, 复旦大学, 2009
[27] Jochen R, Xiao P G, Rudolf B. J Ethnopharmacol, 2001, 75: 133
[28] Chen Z N, Xu P J. Acta Pharmaceutica Sinica, 1993, 28(11): 876
陈泽乃, 徐佩娟. 药学学报, 1993, 28(11): 876
[29] Molina-Torres J, Salazar-Cabrera C, Armenta-Salinas C, et al. J Agric Food Chem. 2004, 52: 4700
猜你喜欢
舰船科学技术(2022年10期)2022-06-17
作文周刊·小学六年级版(2021年20期)2021-05-16
宝藏(2020年1期)2020-10-14
佳木斯大学学报(自然科学版)(2020年1期)2020-02-28
电子制作(2019年20期)2019-12-04
东坡赤壁诗词(2019年4期)2019-09-12
中成药(2017年12期)2018-01-19
中成药(2017年8期)2017-11-22
农产品加工(2017年6期)2017-05-09
电力建设(2015年2期)2015-07-12
