
基于分子络合的分散液液微萃取与高效液相色谱联用检测环境水样中2种苯氧羧酸类除草剂
2018-05-28 07:36张吉苹蒋新娣
色谱 2018年5期
张吉苹, 蒋新娣, 黄 薇, 秦 倩, 周 乔
(江苏瑞达环保科技有限公司, 江苏 盐城 224400)
麦草畏和2,4-二氯苯氧乙酸(2,4-D酸)作为农业上广泛使用的除草剂,是农业经济发展的重要推动力。但是由于它们水溶性较好,可进入农业生态体系,导致地表水和地下水的污染,实现水中该类除草剂的富集与检测对保证水质安全和人类健康具有重要意义。目前已报道的前处理方法有固相萃取(SPE)[1,2]、液液萃取[3]、固相微萃取[4,5]等,这些方法多存在耗时费力或富集倍数小等缺点。分散液液微萃取(DLLME)具有操作简单、快速、成本低、回收率和富集倍数高等诸多优点,被广泛应用于环境样品中有机物的分析[6]。传统DLLME方法常用的萃取剂为氯苯、氯仿、四氯化碳等弱极性或非极性的有机溶剂,其与目标化合物之间基于“相似相溶”原理,因此适用于疏水性化合物的分析,在极性较大的亲水性化合物方面的应用则受到限制。基于分子络合原理的新型分散液液微萃取可利用萃取剂与目标化合物之间的化学反应实现对目标化合物的萃取[7]。目前已报道的极性有机物的DLLME方法常用的萃取剂有磷酸三丁酯(TBP)、碳酸二甲酯(DMC)、碳酸二乙酯(DEC)、三辛胺(TOA)等,它们可以与极性较大的酚类、羧酸类化合物形成稳定且可逆的分子间氢键,从而实现对目标化合物的高效萃取[8]。2010年,Hu等[7]提出了基于分子络合的DLLME,以TBP作为萃取剂成功地检测了环境水样中的4种苯酚类化合物,LOD为7~29 mg/L; 2013年,Teresa Pena等[9]用TBP作为萃取剂检测了环境水样中的苯并三唑和苯并噻唑类化合物,取得了较好的效果。本文以TBP为萃取剂,将基于分子络合的DLLME方法与HPLC联用,实现了水样中两种酸性除草剂的富集和检测。
1 实验部分
1.1 仪器与试剂
Agilent 1220系列高效液相色谱仪,配备二元高压泵、六通阀、恒温箱、紫外检测器、手动进样器和定量阀,购于美国Agilent公司。
去离子水(电导率≥18 MΩ5cm)产自实验室超纯水系统(上海和泰有限公司纯水机Smart-D系列)。TBP、DEC、十一醇、氯化钠、磷酸、氢氧化钾、浓盐酸购于上海国药集团化学试剂有限公司,均为分析纯。麦草畏、2,4-D酸标准品为优级纯,购于德国Sigma-Aldrich公司。色谱纯甲醇购于德国默克公司。
准确称取2种标准品各0.010 0 g,用甲醇溶解,配制成100 mg/L的标准储备液,于4 ℃密封保存,使用时用去离子水稀释至所需浓度。
1.2 色谱条件
EC-C18色谱柱(100 mm×4.6 mm, 4 μm,美国Agilent公司),流动相为甲醇-磷酸缓冲溶液(pH=2.5)(58∶42, v/v),流速为0.8 mL/min,进样体积为20 μL,检测波长为230 nm。
1.3 萃取过程
DLLME方法以TBP作为萃取剂,甲醇作为分散剂,萃取过程如下:取10 mL经硫酸调节pH为0~1.0的水样置于15 mL塑料离心管中,向水样中加入100 μL 100 mg/L的2种目标化合物的混合标准溶液,然后快速向水样中加入100 μL TBP与1 000 μL甲醇的混合液,振摇30 s后进行离心,离心后取出上层油相于1.5 mL微型离心管中,加入80 μL pH为12.0的KOH溶液,超声3 min进行反萃,反萃后以4 000 r/min的转速离心3 min,用移液器取出下层水相40 μL,加入0.05 mol/L的HCl溶液10 μL调节其酸度,最后用微量注射器注入HPLC进行分析。
1.4 萃取率(ER)和富集倍数(EF)的计算
萃取率(ER)按公式(1)进行计算:
(1)
其中n1为萃取后有机相中分析物的物质的量,n2为水样中添加的分析物的物质的量。
富集倍数(EF)按公式(2)进行计算:
(2)
其中c1为萃取后有机相中分析物的浓度,c2为初始水样中分析物的加标浓度。
2 结果与讨论
2.1 实验参数的优化
对影响该方法的萃取效率的因素,如水样的pH值、萃取剂的种类和体积、分散剂的种类和用量、反萃液的pH值和反萃液用量、盐浓度进行了考察。采用萃取率评价萃取效果[10],样品的加标水平设置为1 mg/L,测定结果采用3次测定的平均值。
2.1.1萃取剂的种类
基于分子络合的DLLME方法的萃取剂有TBP、DEC、DMC、三辛基氧膦(TOPO)等含氢键受体的有机物[11]。其中DMC在水中溶解度过大,TOPO为固体,本实验选择TBP、DEC作为萃取剂,并与DLLME方法常用的萃取剂十一醇进行了萃取效果的对比。由图1可看出,TBP获得的萃取率远高于DEC和十一醇,因此选择TBP作为萃取剂。

图 1 萃取剂种类对萃取率的影响(n=3)Fig. 1 Effect of the type of extraction solvent on extraction recovery (ER) (n=3) 2,4-D: 2,4-dichlorophenoxyacetic acid; TBP: tri-n-butyl-phosphate; DEC: diethyl carbonate. Conditions: sample volume, 10 mL; pH 0; extraction solvent volume, 100 μL; methanol volume, 1 000 μL; KOH volume, 80 μL.

图 2 萃取剂体积对萃取率的影响(n=3)Fig. 2 Effect of the volume of extraction solvent on ER (n=3) Conditions: sample volume, 10 mL; pH 0; methanol volume, 1 000 μL; KOH volume, 80 μL.
2.1.2萃取剂的体积
本实验萃取过程包含反萃,所以萃取剂的用量应能保证目标化合物可被完全萃取。向10 mL水样中分别加入40、60、80、100和120 μL TBP,萃取率与萃取剂体积的关系如图2所示,TBP的体积从40 μL到120 μL,萃取率无明显变化。但萃取剂体积小于40 μL时,后续取出全部萃取剂用于反萃的这一步骤所引起的误差将会变大;另一方面,若萃取剂用量过大,反萃时取出全部萃取剂与碱液混合这一过程则会因油水比例过大而引起严重的乳化现象,导致有机相与水相无法彻底分离。因此,综合考虑误差和稀释效应,实验选择TBP的体积为100 μL。
2.1.3分散剂的种类和体积
将甲醇、丙酮和四氢呋喃(THF)作为分散剂,考察其对萃取率的影响。向10 mL水样中加入100 μL TBP与1 000 μL分散剂的混合液。其中,THF与TBP的混合液加入水相中经振摇和离心后获得的有机相体积约为180 μL,说明部分THF离心后仍溶于有机相TBP中,在反萃过程中该有机相与碱液接触,其中部分THF可能会再次溶于碱液中,导致反萃水相体积变化而引起较大的实验误差。因此实验最终考察了甲醇和丙酮两种分散剂,并与不使用分散剂时的萃取率进行对比,分散剂种类与萃取率的关系如图3所示,其中甲醇作为分散剂时的萃取率最高,因此后续实验选择甲醇作为分散剂。

图 3 分散剂种类对萃取率的影响(n=3)Fig. 3 Effect of the type of dispersive solvent on ER (n=3) Conditions: sample volume, 10 mL; pH 0; TBP volume, 100 μL; dispersive solvent volume, 1 000 μL; KOH volume, 80 μL.

图 4 分散剂体积对萃取率的影响(n=3)Fig. 4 Effect of the volume of dispersive solvent on ER (n=3) Conditions: sample volume, 10 mL; pH 0; TBP volume, 100 μL; dispersive solvent volume, 100-1 000 μL; KOH volume, 80 μL.
分散剂甲醇的体积考察范围为100~1 000 μL,甲醇体积与萃取率的关系如图4所示,甲醇体积为1 000 μL时的萃取率最大,此时2种目标化合物的萃取率接近85%和90%。
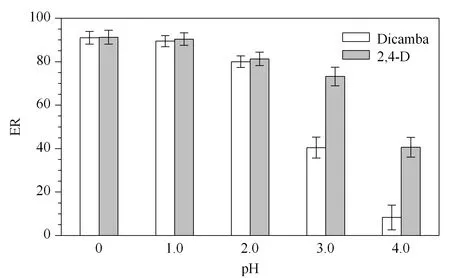
图 5 pH值对萃取率的影响(n=3)Fig. 5 Effect of pH on ER (n=3) Conditions: sample volume, 10 mL; TBP volume, 100 μL; dispersive solvent volume, 1 000 μL; KOH volume, 80 μL.
2.1.4pH值和盐效应
由于麦草畏和2,4-D酸的pKa值分别为1.97和2.6,需将水样的pH值调节至1.0以下才有助于其萃取[12]。为避免调酸所用无机酸与TBP发生络合反应而消耗部分TBP,本实验选择与TBP络合能力相对较弱的硫酸来调节水样的pH值。水样pH为0、1.0、2.0、3.0、4.0时的实验结果如图5所示,pH等于0和1.0时的萃取率相当,pH等于2.0时,萃取率有所下降,至pH等于3.0~4.0时,麦草畏的萃取率明显下降,2,4-D的萃取率缓慢下降。因此本实验选择的最佳pH条件为0~1.0。向10 mL水样中加入0~2.0 g NaCl以考察盐浓度对萃取率的影响,结果发现盐浓度的改变对萃取率无明显影响,因此实验最终选择不加盐。
2.1.5反萃溶液的pH值
在反萃溶液的体积保持80 μL不变的情况下,考察反萃溶液的pH范围为8.0~13.0时pH与萃取率的关系,如图6所示,pH为8.0~10.0时萃取率很低,pH为12.0~13.0时萃取率可达90%,但pH为13.0时,后续调酸步骤需加入的盐酸体积也需相应增大,因此实验最终选择反萃液的pH为12.0。反萃虽然增加了萃取过程的操作步骤,但其可以在很大程度上净化基质,从而提高DLLME方法的选择性。
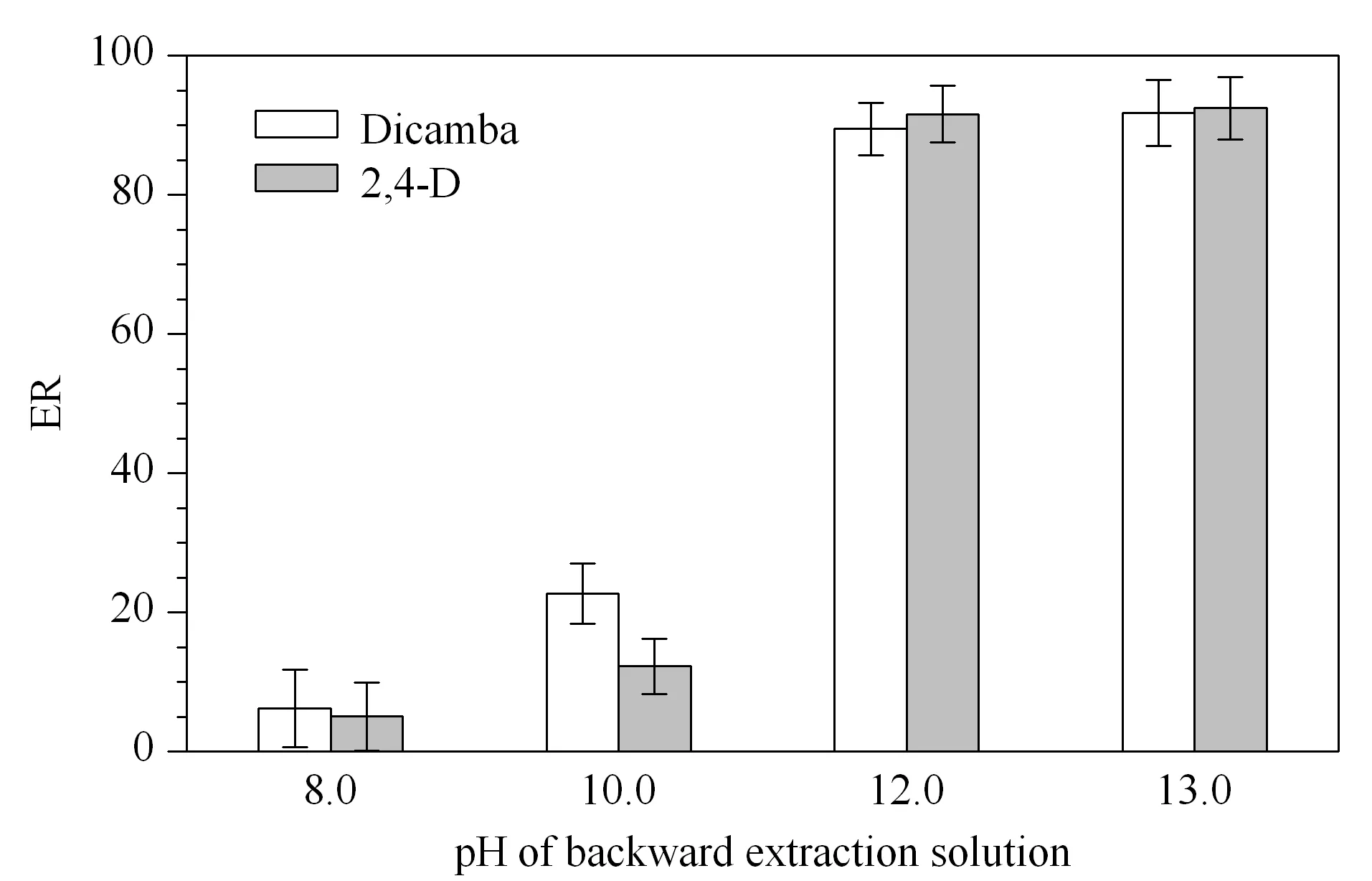
图 6 反萃液pH值对萃取率的影响(n=3)Fig. 6 Effect of pH of backward extraction solution on ER (n=3) Conditions: sample volume, 10 mL; pH 0; TBP volume, 100 μL; dispersive solvent volume, 1 000 μL; KOH volume, 80 μL.
2.1.6反萃液的体积
选择合适的反萃液体积一是为了保证反萃完全,二是为了确保进样体积足够,体积过小不利于进样,过大则相当于稀释从而降低富集倍数。反萃溶液体积考察范围为60~120 μL,结果表明反萃液体积在60~120 μL之间变化时,ER无明显变化,本实验最终选择反萃溶液的体积为80 μL,进样前用20 μL 0.05 mol/L的HCl调节其酸度。
2.2 方法评价
2.2.1方法学考察
在最佳实验条件下(水样10 mL, pH=0~1.0, TBP 100 μL,甲醇1 000 μL, 0.01 mol/L KOH 80 μL),所建方法的富集倍数、检出限(LOD,S/N=3)、日内结果的RSD和日间结果的RSD(对1 mg/L 2种酸性除草剂标准溶液在日内和日间分别平行测定3次所得)见表1, 3种目标化合物在0.5~1 000 μg/L内均具有良好的线性,相关系数(r)不小于0.998 5。

表 1 2种目标化合物的线性范围、相关系数(r)、富集倍数、精密度(n=3)和检出限
*:S/N=3.
2.2.2实际样品分析及回收率试验
从阜宁排污口河道处采集水样,用0.45 μm过滤膜过滤,由所建方法萃取后注入HPLC进行分析,结果显示没有目标化合物被检出。对该实际水样进行 2种酸性除草剂高、中、低3个水平(分别为1.0、5.0和10 μg/L)的加标回收试验,空白水样及部分加标水样的色谱图见图7,样品的加标回收率及RSD分别见表2。
2.2.3与其他方法的对比
本文所建前处理方法与其他已报道的测定烷基苯酚类目标化合物的方法在萃取剂的用量、萃取时间、检出限等方面的对比见表3。可以看出,本文所建方法在萃取时间和操作简便程度方面与SPE、SPME和LLE等前处理方法相比具有明显的优势,且回收率与之相当。

图 7 实际水样和加标水样的色谱图Fig. 7 Chromatograms of a real water sample and spiked real water samples a. a non-spiked real water sample; b. a real water sample spiked with 1.0 μg/L of the two target analytes; c. a real water sample spiked with 5.0 μg/L of the two target analytes.

AnalyteBackground/(μg/L)1.0μg/LR/%RSD/%5.0μg/LR/%RSD/%10.0μg/LR/%RSD/%DicambaND75.77.2185.011.2102.16.862,4-DND75.911.587.78.86104.07.33
ND: not detected. R: spiked recovery.
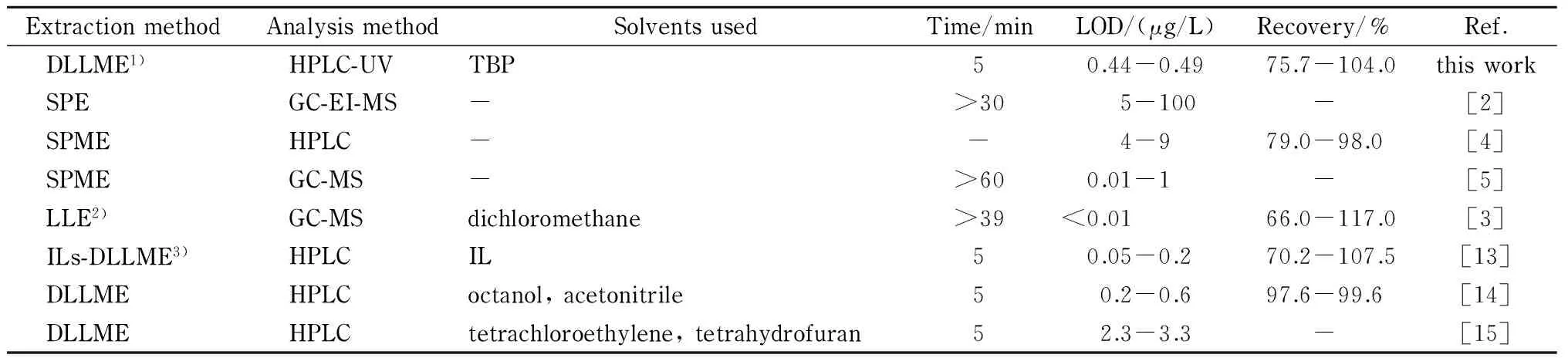
表 3 本文所建方法与其他测定酸性除草剂类目标化合物的前处理方法的比较
1) DLLME: dispersive liquid-liquid microextraction; 2) LLE: liquid-liquid microextraction; 3) ILs-DLLME: ionic liquid dispersive liquid-liquid microextraction.
与表3中3种DLLME方法相比,2,4-D的富集倍数和检出限相当,但基于分子络合的分散液液微萃取在极性更大的目标物的萃取富集方面具有潜在优势,且所用萃取剂低毒,符合绿色化学发展趋势。
3 结论
本文建立了基于络合反应的新型分散液液微萃取前处理方法,与HPLC联用检测环境水样中的2种苯氧羧酸类除草剂,方法应用于实际环境水样,具有较低的检出限和较高的回收率,证明了TBP作为DLLME萃取剂并可应用于麦草畏和2,4-D酸的适用性,为环境水样中苯氧羧酸类除草剂的富集与检测提供了新的对环境友好的前处理方法。
:
[1] Butz S, Heberer T, Stan H J. J Chromatogr A, 1994, 677(1): 63
[2] Maloschik E, Mortl M, Székács A. Anal Bioanal Chem, 2010, 397(2): 537
[3] Zhang L, Gui J Y, Zhang Y T, et al. Physical Testing and Chemical Analysis (Part B: Chem Anal), 2011, 47(1): 8
张莉, 桂建业, 张永涛, 等. 理化检验(化学分册), 2011, 47(1): 8
[4] Wang J B, Wu F L, Zhao Q. Chinese Journal of Chromatography, 2015, 33(8): 849
王家斌, 吴芳玲, 赵琦. 色谱, 2015, 33(8): 849
[5] Scheyer A, Briand O, Morville S, et al. Anal Bioanal Chem, 2007, 387(1): 359
[6] Hashemi B, Zohrabi P, Kim K H, et al. TrAC-Trends Anal Chem, 2017, 97: 83
[7] Hu X Z, Wu J H, Feng Y Q. J Chromatogr A, 2010, 1217(45): 7010
[8] Fan Y Y, Hu S B, Liu S H. J Sep Sci, 2014, 37(24): 3662
[9] Teresa Pena M, Vecino-Bello X, Carmen Casais M, et al. Anal Bioanal Chem, 2012, 402: 1679
[10] Bai X H, Hu S, Chen X. Liquid-Phase Micro-extraction. 1st ed. Beijing: Chemical Industry Press, 2013
白小红, 胡爽, 陈璇. 液相微萃取. 1版. 北京: 化学工业出版社, 2013
[11] Dai Y Y, Qin W, Zhang J, et al. Complex Extraction Chemistry of Organic Compounds. 2nd ed. Beijing: Chemical Industry Press, 2015
戴猷元, 秦炜, 张瑾, 等. 有机物络合萃取化学. 2版. 北京: 化学工业出版社, 2015
[12] Lin C Y, Huang S D. J Chromatogr A, 2008, 1193: 79
[13] Yang S P, Guo Z F, Liu M, et al. Chinese Journal of Analytical Chemistry, 2015, 43(6): 904
杨素萍, 郭振福, 刘敏, 等. 分析化学, 2015, 43(6): 904
[14] Behbahani M, Najafi F, Bagheri S, et al. Environ Monit Assess, 2014, 186: 2609
[15] Melwanki M B, Fuh M R. J Chromatogr A, 2008, 1207: 24
猜你喜欢
理化检验-化学分册(2020年5期)2020-06-15
分析化学(2019年3期)2019-03-30
今日农业(2019年15期)2019-01-03
现代园艺(2017年19期)2018-01-19
天津造纸(2016年1期)2017-01-15
天津科技大学学报(2016年1期)2016-02-28
岩矿测试(2015年3期)2015-12-21
山东工业技术(2015年21期)2015-11-04
橡胶工业(2015年9期)2015-08-29
营销界(2015年23期)2015-02-28
