
18q21杂合缺失致婴儿肝内胆汁淤积症伴智能发育落后1例病例报告
2018-05-28 08:06翟丽娟王能里龚敬宇王建设
中国循证儿科杂志 2018年2期
翟丽娟 杜 鹃 王能里 龚敬宇 王建设
1 病例资料
患儿男,3月2 d,因“皮肤巩膜黄染2月余” 于2014年9月收入复旦大学附属金山医院(我院)儿科病房。
患儿系G1P1,足月顺产,出生体重2 400 g,否认窒息、缺氧或抢救病史。生后因“气促发绀”转入当地医院新生儿科,予吸氧、抗感染等治疗1周。出院第3 d发现皮肤巩膜黄染并逐渐加重,伴尿色黄、粪便颜色浅黄。26日龄在当地医院经皮测胆红素359 μmol·L-1,予“茵栀黄”口服,黄疸减轻。2月龄时因皮肤巩膜仍有黄染再次于当地住院,查肝炎病毒系列、HIV、TORCH近期感染或活动感染指标均为阴性,血串联质谱和尿气相色谱检查均未见明显异常,甲状腺功能正常,给予“熊去氧胆酸(UDCA)、复方甘草酸苷、丁二磺酸腺苷蛋氨酸、茵栀黄”保肝利胆等治疗,患儿皮肤黄染未见明显减轻,遂转至我院就诊。患儿生后前2周为母乳喂养,后改为人工喂养。
患儿母亲孕期无重大疾病、射线暴露或药物服用史。父母非近亲结婚、无遗传代谢性疾病和肝胆疾病家族史。
入院查体:体重4 kg( 实验室常规检查:血常规、凝血功能、空腹血糖正常。表1显示肝功能指标,血清总胆红素(TB)明显升高,以直接胆红素(DB)升高为主,ALT、AST、总胆汁酸(TBA)、甲胎蛋白(AFP)升高,谷氨酰转肽酶(GGT)和白蛋白(ALB)在正常值范围,提示为低GGT胆汁淤积症。 表1 患儿肝功能指标 注 1): GGT(U·L-1)参考值范围:0~4月7~100,~7月5~45,>7月5~25 染色体和基因检查:经患儿父母知情同意后,抽取患儿外周静脉血2 mL,提取基因组DNA,进行染色体芯片检测(美国Affymetrix 公司Agilent aCGH芯片),使用Affymetrix Chromosome Analysis Suite(ChAS)软件[版本:2.0.0.195(r5758) ]进行数据处理和分析。发现18号染色体长臂(18q21.2 - q21.33)11.6Mb的缺失(chr18:49,703,557-61,366,422)和8号染色体短臂(8p23.2)961Kb的缺失(chr8:5,118,481-6,079,790),经查阅Decipher数据库,发现8号染色体缺失区域内不包含目前已知的基因,18号染色体长臂缺失区域包含ATP8B1和TCF4基因。以患儿白细胞DNA为模板,PCR扩增ATP8B1基因(ENSG00000283684)编码区27个外显子及其侧翼序列,扩增及测序引物,PCR产物经明码(上海)生物科技有限公司测序,应用BWA软件(版本:0.7.9a)比对测序结果,在编码区发现2个SNP,分别是第8外显子的c.696T>C (p.D232D)和第10外显子的c.811A>C(p.R271R),通过查询Mutationtaster软件预测为非致病性。 ATP8B1基因位于染色体18q21-22区域,全长约176.68 kb,含28个外显子,编码ATP8B1蛋白,又称为FIC1蛋白[2,3]。FIC1蛋白是P型ATP酶4型亚家族的成员,为多重跨膜蛋白,表达于上皮细胞的顶膜,包括肝细胞的毛细胆管膜[4,5]。ATP8B1突变可导致进行性家族性肝内胆汁淤积症(PFIC)1型、良性复发型肝内胆汁淤积症(BRIC)1型和妊娠期肝内胆汁淤积症(ICP)1型和暂时性婴儿胆汁淤积症。已有多项研究证实,ATP8B1是一种内翻转酶,介导氨基磷脂和磷脂酰丝氨酸由细胞外膜向内膜的内转位,但其引起胆汁淤积的机制尚不完全清楚[6~8]。ATP8B1缺陷病的临床表型包括PFIC 1型、BRIC 1型、ICP 1型及暂时性婴儿胆汁淤积症,是一个轻重不等的谱系疾病。表现为PFIC1型者,黄疸持续,并存在进行性的肝脏损伤,可在儿童早期迅速进展至终末期肝病,如不进行肝移植手术,可能会在10或20年内死亡[9]。表现为BRIC1型者,可反复出现胆汁淤积,能自发缓解,不遗留严重的肝脏损害,首次黄疸可出现于1~50岁,多数在20岁之前发病[10],通常会有2~4周以乏力、食欲减退和瘙痒为特征的黄疸前期,无明显的发作诱因,发作持续时间和次数个体差异较大,可持续1~18个月,以2~3个月常见。ICP1型以孕期瘙痒及血清胆汁酸水平增高为特征,可发生于PFIC 1型和BRIC 1型家系中。暂时性婴儿胆汁淤积症仅表现为婴儿期暂时性的胆汁淤积,其表现和生化特征与BRIC1型发作期相似。PFIC型常为ATP8B1基因严重的(插入、缺失、无义或剪切突变)纯合或复合杂合突变,导致效应蛋白极少或检测不到;BRIC型则为ATP8B1基因杂合突变或对蛋白功能损伤相对较轻的纯合突变[9,11],PFIC 1型、BRIC 1型、ICP 1型及暂时性婴儿胆汁淤积症均是因ATP8B1基因突变所致,但BRIC 1型及暂时性婴儿胆汁淤积症的临床表现及预后均较PFIC 1型轻,因此临床上认为BRIC 1型及暂时性婴儿胆汁淤积症是PFIC1型的良性表现形式,但部分开始表现为BRIC的患者反复发作,可进展为PFIC型。不论PFIC型,还是BRIC型或暂时性婴儿胆汁淤积的发作期,ATP8B1缺陷病的实验室检查的共同特点为血清胆汁酸和胆红素水平升高,GGT水平正常[12]。 本文报告的病例于新生儿期出现胆汁淤积症,外院多次嗜肝病毒及非嗜肝病毒血清标志物检查均阴性,故不考虑宫内感染致胆汁淤积的可能;血串联质谱、尿气相色谱、空腹血糖均正常,且GGT水平始终在正常值范围,因此排除了先天性胆道闭锁、Alagille综合征及Citrin缺乏症等;血清生化示转氨酶、总胆汁酸、甲胎蛋白均升高,排除了胆汁酸合成缺陷。染色体芯片检测显示,患儿18号染色体长臂(18q21.2 - q21.33)和8号染色体短臂(8p23.2)缺失,18号染色体缺失区域内包含了ATP8B1基因。对另一条同源染色体上ATP8B1等位基因全部编码外显子进行测序,发现2处SNP,均表现为单一峰,和患儿的整个ATP8B1基因存在杂合缺失相符。ATP8B1缺陷可导致持续低GGT的肝内胆汁淤积症,这与该患儿的临床生化指标一致,因此认为该患儿出现的肝内胆汁淤积与18号染色体杂合缺失导致ATP8B1基因缺陷有关。该患儿经UDCA利胆、苯巴比妥诱导肝酶、补充脂溶性维生素等治疗,1岁龄时黄疸消退,肝功能各项指标恢复正常,其病程的演变符合暂时性婴儿胆汁淤积症。随访至2岁10个月,患儿再未出现黄疸,胆汁淤积未再复发,但其远期是否会发展为BRIC型,仍有待继续随访。 本例患儿入院时无明显特殊面容,随着年龄增长,逐渐出现特殊面容(嘴宽大、唇厚、鼻梁宽而高、鼻尖突出、下颌略微前突),患儿身高和体重均低于同龄同性别幼儿,有明显的智力和语言发育障碍,还有小头畸形、无意识的手部动作、严重便秘。ATP8B1缺陷并无特殊面容和智能发育障碍的相关报道,进一步查阅Decipher数据库,发现18号染色体缺失区域内还包含TCF4基因。TCF4基因位于染色体18q21[13],全长约360 kb,含20个外显子,编码一种碱性螺旋-环-螺旋(bHLH)转录因子,属于E蛋白质家族,表达于支气管周围、肾间质、交感及副交感神经、小肠神经节、胚胎脏壁层间质细胞,特别是中枢神经系统(包括端脑、间脑、小脑[14,15]),其在中枢神经系统的发育中起转录调控作用。TCF4基因突变可导致皮特-霍普金斯综合征(PHS)。 ATP8B1缺陷病发病率低,PHS在国内更是鲜有报道。本文报告的病例为染色体18q21杂合缺失导致的ATP8B1缺陷病(婴儿肝内胆汁淤积症)合并PHS。本文于2018年3月1日分别检索了中国知网[( 摘要=ATP8B1 并且 摘要=皮特-霍普金斯 ) (模糊匹配);( 摘要=ATP8B1 并且 摘要=Pitt-Hopkins ) (模糊匹配)]、万方数据知识服务平台[摘要:(∷ATP8B1∷)*摘要:(∷皮特-霍普金斯∷);摘要:(∷ATP8B1∷)*摘要:(∷Pitt-Hopkins∷)]均未检索到ATP8B1缺陷病合并PHS的病例;同时,在Google scholar(“ATP8B1” and “Pitt-Hopkins Syndrome”)检索到4篇可能相关的文献,阅读摘要后发现3篇不相关。法国学者Jacquemin在2010年首次报告了1例18q21缺失导致的ATP8B1缺陷病合并PHS的病例[25],当时认为这两个疾病之间可能是偶然的联系,而本文患儿亦为ATP8B1缺陷病合并PHS,提示这两种疾病的发生之间可能存在一定的关联,当发现婴儿胆汁淤积或PHS时应同时检测这两个基因。 综上所述,本文采用染色体芯片技术和基因测序确诊了1例婴儿期肝内胆汁淤积症合并PHS病例,18q21缺失区域内包含ATP8B1及TCF4基因,主要临床表型为胆汁淤积、智能发育落后和特殊面容等。提示临床上对于原因不明的胆汁淤积,应重视分子学诊断。本例患儿,用常规的基因外显子测序技术检测ATP8B1基因全部外显子时,未发现致病性基因突变,而染色体芯片检测发现了染色体片段缺失。因此,常规的基因外显子测序技术可能会漏诊一些染色体片段缺失的病例,联合使用染色体芯片技术则可以弥补其不足。 参考文献 [1] 首都儿科研究所, 九市儿童体格发育调查协作组. 中国七岁以下儿童体重、身长/身高和头围的生长标准值及标准化生长曲线. 中华儿科杂志, 2009, 47: 173-173 [2] Gonzales E, Spraul A, Jacquemin E. Clinical utility gene card for: Progressive familial intrahepatic cholestasis type 1. Eur J Hum Genet, 2014, 22(4). doi: 10.1038/ejhg. 2013. 186 [3] Bull LN, van Eijk MJ, Pawlikowska L, et al. A gene encoding a P-type ATPase mutated in two forms of hereditary cholestasis. Nat Genet, 1998. 18(3): 219-224 [4] Folmer DE, Elferink RP, Paulusma CC. P4 ATPases-lipid flippases and their role in disease. Biochim Biophys Acta, 2009, 1791(7): 628-635 [5] Holthuis JC, Levine TP. Lipid traffic: floppy drives and a superhighway. Nat Rev Mol Cell Biol, 2005, 6(3): 209-220 [6] Ujhazy P, Ortiz D, Misra S, et al. Familial intrahepatic cholestasis 1: studies of localization and function. Hepatology, 2001, 34 (4 Pt 1): 768-775 [7] Paulusma CC, Folmer DE, Ho-Mok KS, et al. ATP8B1 requires an accessory protein for endoplasmic reticulum exit and plasma membrane lipid flippase activity. Hepatology, 2008, 47(1): 268-278 [8] Cai SY, Gautam S, Nguyen T, et al. ATP8B1 deficiency disrupts the bile canalicular membrane bilayer structure in hepatocytes, but FXR expression and activity are maintained. Gastroenterology, 2009, 136(3): 1060-1069 [9] 李丽婷, 王建设. ATP8B1缺陷病. 肝脏, 2012, 17(8): 581-583 [10] Jansen PL, Sturm E. Genetic cholestasis, causes and consequences for hepatobiliary transport. Liver Int, 2003, 23(5): 315-322 [11] 贺希, 苏海滨, 刘振文, 等. 良性复发性肝内胆汁淤积1例. 肝脏, 2013, 18(2): 139-140 [12] Bijleveld CM, Vonk RJ, Kuipers F, et al. Benign recurrent intrahepatic cholestasis: altered bile acid metabolism. Gastroenterology, 1989, 97(2): 427-432 [13] Amiel J, Rio M, Redon R, et al. Mutations in TCF4, encoding a class I basic helix-loop-helix transcription factor, are responsible for Pitt-Hopkins syndrome, a severe epileptic encephalopathy associated with autonomic dysfunction. Am J Hum Genet,2007, 80(5): 988-993 [14] Zhuang Y, Cheng P, Weintraub H. B-lymphocyte development is regulated by the combined dosage of three basic helix-loop-helix genes, E2A, E2-2, and HEB. Mol Cell Biol, 1996, 16(6): 2898-2905 [15] Navarrete K, Pedroso I, De Jong S, et al. TCF4 (e2-2, ITF2): A schizophrenia-associated gene with pleiotropic effects on human disease. Am J Med Genet B Neuropsychiatr Genet, 2013, 162B(1): 1-16 [16] Pitt D, Hopkins I. A syndrome of mental retardation, wide mouth and intermittent overbreathing. Aust Paediatr J, 1978, 14(3): 182-184 [17] Ardinger HH, Welsh HI, Saunders CJ. Pitt-Hopkins Syndrome. In: Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP (eds). GeneReviews [Internet]. University of Washington: Seattle, WA, USA, 1993-2012. Aug 30, 2012 [18] Takano K, Tan WH, Irons MB, et al. Pitt-Hopkins syndrome should be in the differential diagnosis for males presenting with an ATR-X phenotype. Clin Genet, 2011, 80(6): 600-601 [19] Marangi G, Ricciardi S, Orteschi D, et al. The Pitt-Hopkins syndrome: report of 16 new patients and clinical diagnostic criteria. Am J Med Genet A, 2011,155A(7): 1536-1545 [20] Peippo MM, Simola KO, Valanne LK, et al. Pitt-Hopkins syndrome in two patients and further definition of the phenotype. Clin Dysmorphol, 2006, 15(2): 47-54 [21] de Pontual L, Mathieu Y, Golzio C, et al. Mutational, functional, and expression studies of the TCF4 gene in Pitt-Hopkins syndrome. Hum Mutat, 2009, 30(4): 669-676 [22] Irina G, Chantal M, Pierre C, et al. TCF4 deletions in Pitt-Hopkins syndrome. Hum Mutat, 2008, 29(11): E242-E251 [23] Marangi G, Ricciardi S, Orteschi D, et al. Proposal of a clinical score for the molecular test for Pitt-Hopkins syndrome. Am J Med Genet A, 2012, 158A(7): 1604-1611 [24] Lehalle D, Williams C, Siu VM, et al. Fetal pads as a clue to the diagnosis of Pitt-Hopkins syndrome. Am J Med Genet A, 2011, 155A(7): 1685-1689 [25] Jacquemin E, Malan V, Rio M, et al. Heterozygous FIC1 deficiency: a new genetic predisposition to transient neonatal cholestasis. J Pediatr Gastroenterol Nutr, 2010, 50(4): 447-449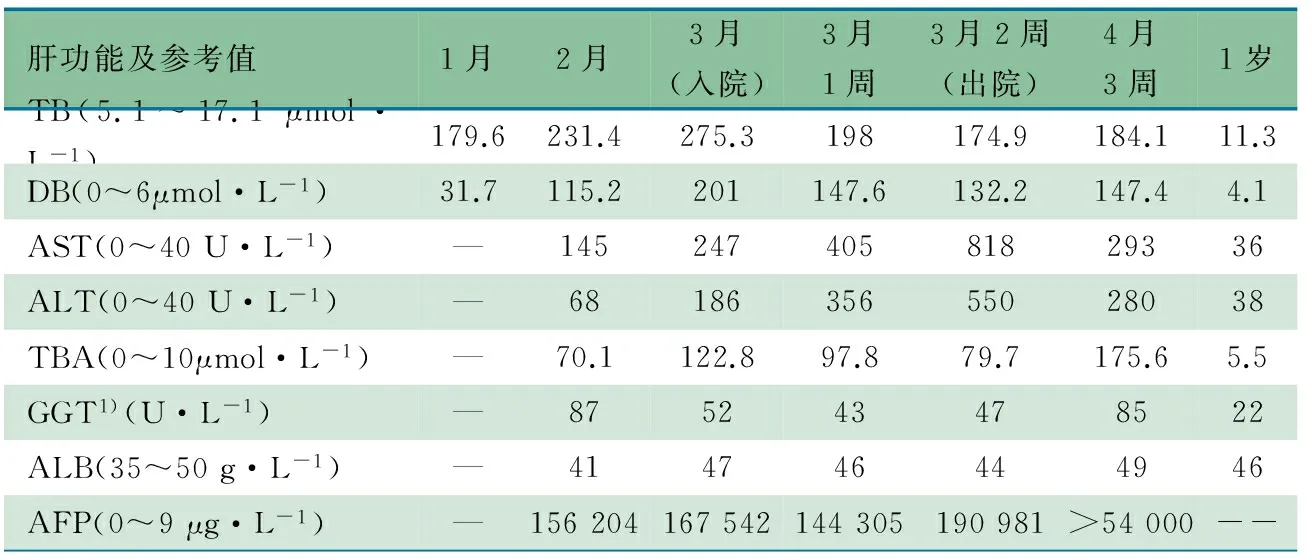

2 讨论
猜你喜欢
今日畜牧兽医(2022年7期)2023-01-05
基层中医药(2022年7期)2022-11-17
保健医苑(2022年5期)2022-06-10
种子(2021年3期)2021-04-12
中国临床医学影像杂志(2019年6期)2019-08-27
浙江中医杂志(2019年3期)2019-01-05
祝您健康·文摘版(2018年6期)2018-10-21
中国医药指南(2017年3期)2017-11-13
校园英语·下旬(2017年7期)2017-07-14
科技视界(2016年27期)2017-03-14
