
In fluence of Zr,Ce,and La on Co3O4 catalyst for CO2 methanation at low temperature☆
2018-05-25 11:26:23YuwenZhouYuexiuJiangZuzengQinQinruoXieHongbingJi
Yuwen Zhou ,Yuexiu Jiang ,Zuzeng Qin ,2,*,Qinruo Xie ,Hongbing Ji,2,*
1 SchoolofChemistry and ChemicalEngineering,GuangxiKey Laboratory ofPetrochemicalResource Processing and Process Intensi fication Technology,GuangxiUniversity,Nanning 530004,China
2 School of Chemistry,Sun Yat-sen University,Guangzhou 510275,China
3 School of Biology and Chemical Engineering,Guangxi University of Science and Technology,Liuzhou 545006,China
1.Introduction
Over the past centuries,CO2has become the main carbon resource due to the decreases of limited resources such as coal,oil and natural gas[1,2].However,the CO2concentration in the atmosphere has consequently risen,which arguably contributes to the “greenhouse effect”,and increase the global temperatures and climate change[3,4],and it is high time that effective measures should be taken to decrease the emission of CO2.CO2methanation is a simple reaction and can generate methane under atmospheric pressure,and previous reports showed that the Ni catalysts used in the CO2hydrogenation have preferable activity on the CO2conversion and the CH4selectivity;however,the reaction requiresmuch highertemperatures[5–7],such as CO2methanation on Ni/La2O3at 350 °C and 1.5 MPa[8],Ni/HNaUSY at 400 °C[5],and the Ni/MOF at 320°C[6].Furthermore,carbon deposited could easily find on the Ni-based catalysts in a CO2methanation process[9,10],which would lead the inactivation of catalysts.
In the other hand,Co-based catalysts were used in Fischer–Tropsch synthesis,which synthesized the syngas to liquid hydrocarbon,accomplished with a small amount CH4[11,12].Therefore,through adjusting the properties of COx/H2to control the products contribution,and using the Co-based catalyst in the CO2methanation reaction would be an effective catalyst,which was different from the conventional Ni-based catalyst[13,14]and exhibited better resistance to inactivation[15–17].However,the reaction temperature of CO2methanation was still higher,and the CO2conversion and CH4selectivity need to be further improved.At present,adding a promoter to the catalysts would efficiently improve the catalytic activity and reduce the reaction temperature,i.e.,the addition of a second metal(e.g.,Zr,Ce,or La)to the catalysts would obviously promote the metal dispersion,increase the reducibility of active metal,and reduce the crystallite size,which improved the activity of the catalysts[18–22].The Ce addition to Ni increased the dissociation and the hydrogenation activity of CO2and strengthened the interaction between Ce and Ni,resulting to highercatalytic activity of Ce-Ni/Al2O3[23].Mg was added by incipient wetness impregnation and ion exchange and improved the performance of the Ni-zeolite catalysts for CO2methanation;the important improvements of the catalytic performance(CO2conversion and CH4selectivity increased around 20%at 350–450 °C)were also found for the Mg-exchanged 5%Ni/zeolite[21].La,Ce,Pr,Eu&Gd were modi fied to Ni/γ-Al2O3by using aqueous incipient impregnation method[24],and 5%Pr-12%Ni/γ-Al2O3showed the highest CO2conversion of 98.2%with 100%CH4selectivity at 300°C for investigated reaction conditions.
However,the effects of modi fiers on the catalyst properties and the CO2methanation activities on a Co-catalyst were seldom reported.In the present study,based on our previous study of the modi fied Cu-Fe catalysts for CO2hydrogenation to dimethyl ether[18–20],Zr-,Ce-,and La-modi fied Co3O4were prepared via a co-precipitation method and used for the catalytic hydrogenation of CO2to methane at a low temperature of 140–220 °C;the effects of the modi fier type on the catalysts structure and the catalytic activities of CO2methanation were investigated;the stability of the catalysts was also studied.
2.Experimental
2.1.Preparation of catalysts
The Co3O4and Zr-,Ce-,and La-modi fied Co3O4catalysts were prepared via a co-precipitation method.The Co(NO3)2was prepared in the deionized water to a concentration of 0.2 mol·L-1,and based on the ZrO2amount that was 2 wt%of the Co3O4,the Zr(NO3)4was added to the Co(NO3)2aqueous solution to obtain a nitrate aqueous mixed solution.Subsequently,50 ml of Co(NO3)2and Zr(NO3)4mixture aqueous solutions and 0.5 mol·L-1of Na2CO3aqueous solution were added dropwise in the parallel flowing to 100 ml of deionized water at 70 °C until the pH=9,and a 400 r·min-1stiring,and aged for 4.0 h atambienttemperature to obtain the Zr-Co3O4precursor.The precursor was filtered and dried at 110 °C for 12 h,and calcined at 450 °C for 4.0 h.Finally,the 2-wt%Zr modi fied Co3O4powder was grounded to 20–40 meshes for the reaction,which was marked as Zr-Co3O4.The Co3O4,Ce-Co3O4,and La-Co3O4were prepared in the same method.
2.2.Characterization of the catalysts
The X-ray diffraction(XRD)was tested by using a Bruker D8 Advance X-ray diffractometer.The isotherm of nitrogen adsorption and desorption was measured by an ASAP 2000 physical adsorption instrument(Micromeritics Instrument Corp.),the catalyst surface area was calculated via Brunauer–Emmett–Teller(BET)method,and the pore size distribution curve was determined using the Barrett–Joyner–Halenda(BJH)model,which was based on the isotherm of desorption side.A Thermo ESCALAB 250X multifunction imaging electron spectrometer(Thermo Fisher Scientific Co.,Ltd.),which equipped with an Al Kαradiation source,was used to obtain the X-ray photoelectron spectrum(XPS)of catalysts,and the XPS analysis was conducted at 150 W with a pass energy of 40 eV.
Temperature program reduction(H2-TPR)was determined using a DAS-7000 multifunction catalyst analysis system(China Hunan Huasi Technology Co.,Ltd.).The samples(50 mg)were purged with N2(30 ml·min-1)at 300 °C to remove physically adsorbed water followed by cooling to 50°C,and then reduced in a flow of 8%(by volume)H2/Ar(30 ml·min-1)at a heating rate of 10 °C·min-1up to 500 °C.Thermal conductivity detector(TCD)was used to monitor the consumption ofH2.
The CO2-TPD experiments of catalyst samples were taken in a DAS-7000 multifunction catalyst analysis system(China Hunan Huasi Technology Co.,Ltd.).Atypicalsample mass of100 mg was reduced at400°C with an H2(99.999%) flow of30 ml·min-1for 1 h and then cooled to 50°C with 30 ml·min-1of N2.Subsequently,the CO2was introduced at a flow rate of 30 ml·min-1for 1 h at 50 °C,and then the catalysts were purged with 30 ml·min-1of N2for 1 h to remove the physical adsorption of CO2.Until the TCD signal was stabilized,the reactor temperature was programmed to increase ata rate of10°C·min-1to 700 °C,and the amount of CO2in the effluent was measured via TCD and recorded as a function of temperature.
The FTIR of adsorbed pyridine was conducted using a Tensor II FTIR spectrometer(Bruker Corporation),the samples were added into a diffuse sample cell,and the samples were evacuated at 150°C for 1 h to record the background spectrum,and subsequently saturated with pyridine and evacuated at 150°C for 1 h,and the Py-IR spectra were recorded at the spectrum resolution of 4 cm-1after subtracting the sample background.
2.3.CO2 methanation on Co3O4 and Zr-,Ce-,and La-Co3O4
The CO2methanation was carried outin a fixed-bed reactor,consisting of a stainless-steel reaction tube with an 8-mm inner diameter.A 100 mg catalyst was taken into the reactor and reduced at 400°C for 3 h with 40 ml·min-199.999%H2and cooled to the room temperature.Subsequently,the H2and CO2in a 4:1 molar ratio was fed into the reactor at 30 ml·min-1,a gaseous hourly space velocity(GHSV)of18,000 ml·-h-1,and the catalytic hydrogenation ofCO2to CH4was reacted at80–220°C and 0.5 MPa.The gas product amounts were tested by using an online gas chromatograph(Agilent 4890D)equipped with a thermal conductivity detector(TCD),the products of the reaction included CH4,CO,and C2H4at the reaction temperature in the present study,and the CO2conversion(XCO2)and the CH4selectivity(SCH4)were calculated by using Eqs.(1)and(2)based a peak area normalization method.

where,XCO2was the CO2conversion(%);SCH4was the CH4selectivity(%),and the ACO2,ACO,ACH4,and AC2H4were the peak areas of CO2,CO,CH4,and C2H4,respectively.
3.Results and Discussion
3.1.XRD analysis
The XRD patterns of Co3O4,Zr-,Ce-,and La-Co3O4were shown in Fig.1(a).The diffraction peaks at 31.4°,36.9°,45.0°,59.5°,and 65.5°were assigned to the cubic phase Co3O4(JCPDS 65-3103),which existed on all catalysts,and the diffraction peak at 2θ=28.5°in the Ce-Co3O4was attributed to the CeO2phase.Much weakerand broadened Co3O4diffraction peaks were observed on the modi fied catalysts compared to the Co3O4,indicating the crystallite size of Co3O4is smaller after being modified by Zr,Ce,or La.The crystallite sizes of the Co3O4(311)plane in the Co3O4,La-Co3O4,Ce-Co3O4,and Zr-Co3O4calculated by using the Sherrer equation[25]was 23.20,20.21,18.53,and 17.55 nm,respectively,indicating the addition Zr,Ce,and La decreased the crystallite size of Co3O4,which would promote the catalytic activity[26].On the other hand,regarding the XRDpatterns ofthe reduced catalysts in Fig.1,no Co3O4crystalline was observed.The diffraction peaks at 41.7°,44.4°,and 47.1°were the metallic Co-hcp(hexagonal close-packed),indicating the active site for CO2methanation was the metallic Co.In addition,a minor diffraction peak at 75.9°,corresponding to the CoO crystalline,was also observed,which was the partialoxidation ofcobaltwhile in the catalystpreparation process and during sample transfer in the XRD chamber.However,no crystalline Zr or La oxide phase was detected in the modi fied catalysts,which might be attributed to these elements presenting in small quantities or existing in an amorphous state[27].
皮带运输机是矿山生产和开采之中的重要设备,它能节约人力成本,提高矿山生产的效率。皮带运输机以其自身的使用便捷、运输量巨大等优点,在很长一段时间内,都是矿山的重要设备。尤其是在煤矿生产之中,离不开皮带运输机的使用。但在实际应用之中,因为种种因素的影响,皮带运输机也总会发生一些问题影响生产速度,有时还会造成人员伤亡。因此,皮带运输机中必须采用PLC控制技术,保证皮带运输机安全高效运行。
3.2.Nitrogen adsorption/desorption of catalysts
Fig.2 showed the N2adsorption–desorption isotherms and pore size distribution profiles of the Co3O4,Zr-,Ce-,and La-Co3O4,which confirmed all catalysts were mesostructured materials[28].From Table 1,the specific surface areas of Co3O4,Zr-,Ce-,and La-Co3O4was 47,70,65,and 69 m2·g-1,respectively,which indicated the Zr modi fication would increase the dispersion of Co species in the Zr-Co3O4and would provide more activity sites for the CO2catalytic hydrogenation reaction.Furthermore,compared with the Ce-Co3O4and La-Co3O4,a smaller average pore diameter of Zr-Co3O4(16.13 nm)and a narrower pore size distribution[in Fig.2(b)]would provide favorable conditions for the adsorption and activation of CO2molecules[13],which would lead to a high CO2catalytic hydrogenation activity.

Fig.1.XRD patterns of(a)Co3O4,(b)La-Co3O4,(c)Ce-Co3O4,(d)Zr-Co3O4 calcined at 450 °C(a)and after H2-reduced at 400 °C(b).

Fig.2.Nitrogen adsorption/desorption isotherms(a)and pore size distribution profiles(b)of(a)Co3O4,(b)La-Co3O4,(c)Ce-Co3O4,(d)Zr-Co3O4 catalysts.

Table 1 Textural properties of Co3O4,La-Co3O4,Ce-Co3O4,and Zr-Co3O4 catalysts
3.3.H2-TPR analysis
The H2-TPR profiles of the mentioned catalysts were shown in Fig.3,and three Gaussian fitting peaks(α,β,and γ)were shown within 200–450°C.The peak positions and their area were summarized in Table 2.Co3O4was reduced by hydrogen to obtain Co via a two-step reduction:Co3O4→CoO→Co0[13,29].In Fig.3,the low-temperature hydrogen consumption peaks α and β attributed to the reduction of the Co3O4to CoO and the CoO to the metallic cobalt on the catalysts surface,respectively;the peak γ occurred in the Zr,Ce,or La modi fied Co3O4would attribute to the reduction of Co3O4which interacted with ZrO2,CeO2,or La2O3[30].

Fig.3.H2-TPR profiles for(a)Co3O4,(b)La-Co3O4,(c)Ce-Co3O4,(d)Zr-Co3O4 catalysts.The solid curves are experimental curves,and the broken curves are Gaussian multipeak fitting curves.

Table 2 Temperatures and areas of the reduction peaks of Co3O4,La-,Ce-,and Zr-Co3O4 catalysts①
From Table 2,the peak α and β of the Zr-,Ce-,and La-Co3O4were centered at 258 and 250 °C,257 and 318 °C,and 324 and 304 °C,respectively,which were slightly lower than that of the Co3O4,261°C and 324°C,respectively,indicating the reducibility ofthe Co3O4(peakα and β)would improve by the addition of Zr,Ce,and La.Furthermore,the peak γ of Zr-Co3O4was wider than that of the Co3O4,Ce-and La-Co3O4,which suggested a stronger interaction between Co3O4and ZrO2was formed[31,32],and the reduction of the Co-Zr species required a much higher temperature at low Zr amount,leading the Zr-Co3O4reduced difficulty.For the Zr-Co3O4,the ratio of α and β peaks was 1.4,which caused by the weak interaction between cobalt and zirconium,and the Co2+was not completely reduced to Co in the β peak accompanied with the beginning of the reduction in the γ peak;more Co would be reduced in the γ peak;furthermore,some of the cobalt ions would enter the zirconia lattice and formed Co-Zr clusters.In the present study,the Zr-Co3O4was reduced at 400°C,which led to a partial reduction of Co,and some Co2+existed in the catalysts,which might serve as the active site for CO2methanation,resulting in the optimal CO2conversion and CH4selectivity,which agree with the higher CH4selectivity thatwas observed in the Fischer–Tropsch synthesis when Co catalysts were not completely reduced[11,33].
3.4.XPS analysis
To clarify the oxidation states of the elements on the catalysts surface,the samples were characterized by XPS,and the results were shown in Fig.4.
From the Co 2p spectrum of the calcined catalysts in Fig.4(a),the binding energy of Co 2p3/2in the Co3O4was 779.6 eV and 780.2 eV,along with the featured satellite peaks of approximately 789.7 eV,and the binding energy of Co 2p1/2was 794.8 eV and 795.3 eV along with the featured satellite peaks of approximately 804.5 eV,suggesting that Co occurred as the form of Co3+and Co2+in Co3O4[34].After being modi fied with Zr,Ce,and La,the Co 2p3/2and Co 2p1/2peaks red shifted by 0.42,0.21,and 0.18 eV,and 0.45,0.15,and 0.13 eV,respectively,which suggested that the Zr,Ce,and La exchanged electrons with the Co3O4,decreasing the outer-shell electron density of Co and slightly affecting the chemical combination state of Co3O4[35,36].A higher binding energy shift on the Zr-Co3O4compared to the La-and Ce-Co3O4which was attributed to a stronger interaction of Co3O4with ZrO2[32,37],which agreed with the H2-TPRresults.Moreover,from the XPS profile in Fig.4(b),(c),and(d),the Zr,Ce,and La existed in the catalysts as Zr4+,Ce3+/Ce4+,and La3+.Furthermore,the XPS spectra of Co 2p for the four mentioned catalysts reduced at 400°C were shown in Fig.5.Anew Co 2p3/2peak appeared at778.0 eV,attributing to the metallic cobalt[38],which agreed with the XRD results.The intensity and area of this peak increased after the addition of Zr,indicating more metallic Co on the catalyst surface.The peak and the satellite peaks for Co2+were detectable even when the catalyst was reduced at 400°C,which might had been caused by the partial oxidation during the preparation of the sample.

Fig.5.XPS spectra of Co 2p regions for reduced(a)Co3O4,(b)La-Co3O4,(c)Ce-Co3O4,and(d)Zr-Co3O4 catalysts.
3.5.Surface basicity and acidity analysis

Fig.4.Co 2p(a),Zr 3d(b),Ce 3d(c)and La 3d(d)XPS spectra of the Co3O4,La-Co3O4,Ce-Co3O4 and Zr-Co3O4 catalysts.

Fig.6.CO2-TPD profiles(a)and FT-IR spectra of pyridine adsorbed at a desorption temperature of 150°C(b)of the pre-reduced(a)Co3O4,(b)La-Co3O4,(c)Ce-Co3O4,(d)Zr-Co3O4 catalysts.
The surface basicity of the four catalysts was analyzed by CO2-TPD;the CO2-TPD profiles of the pre-reduced Co3O4before and after being modi fied by Zr,Ce,and La were shown in Fig.6(a).Three peaks at 50–200°C,300–400°C,and 500–700 °C were observed in the fourcatalysts,which were assigned to the weak(α peak),medium(β peak)and strong(γ peak)basic sites,respectively[39].After the addition of Zr,the peak area of weak and medium basic sites for Zr-Co3O4exhibited much greater than that of Co3O4,La-,and Ce-Co3O4,indicating the adsorption amount of CO2was significantly improved by adding Zr,and more CO2molecular were activated at 100–200 °C on the Zr-Co3O4,which would increase the catalytic activity.On the contrary,the addition of Ce and La affected slightly on the surface basicity,and the strength of strong basic site decreased after adding Zr or Ce to Co3O4.Therefore,the enhanced basicity by adding Zr to the Co3O4would probably improve the adsorption and activation of CO2on the catalyst surface,resulting in an improvement on the catalytic activity.
3.6.Hydrogenation of CO2 on Co3O4,Zr-,Ce-,and La-Co3O4
After a one-hour reaction at the specified temperature,the catalytic CO2methanation on the Co3O4,and Zr-,Ce-,and La-Co3O4were shown in Fig.7.Obviously,the CO2conversion increased with the increasing reaction temperature from 80 to 220°C on all catalysts.Compared to the Co3O4,the Zr-Co3O4exhibited a higher CO2conversion and CH4selectivity,and the Ce-and La-modi fied Co3O4has insignificant effect on the catalytic activity,suggesting the addition of the Zr can improve the catalytic activity of the Co3O4for CO2methanation.Especially,the Zr-Co3O4exhibited a higher catalytic hydrogenation activity for CO2hydrogenation than the Ce-,and La-Co3O4catalysts,and had higher CH4selectivity under 180–220 °C.For the Co3O4,Ce-,and La-Co3O4,the CO2conversion and CH4selectivity at 200°C were 22.8%,26.1%,and 22.1%and 95.4%,95.3%,and 93.8%,respectively.While the CO2conversion on the Zr-Co3O4increased from 0.84%to 58.2%when the temperature increased from 80 °C to 200 °C,the CH4selectivity was retained at 100%at higher temperatures.Even when the temperature increased to 220°C,the CO2conversion and CH4selectivity was 66.3%and 97.4%,respectively.Therefore,the Zr-Co3O4was the optimal catalysts among the four mentioned catalysts for CO2methanation.In addition,the CO was not detected when reaction temperature was below 200°C;it was detected only when the reaction temperature was higher than 200°C,and the byproducts including small amount of C2H6were detected.
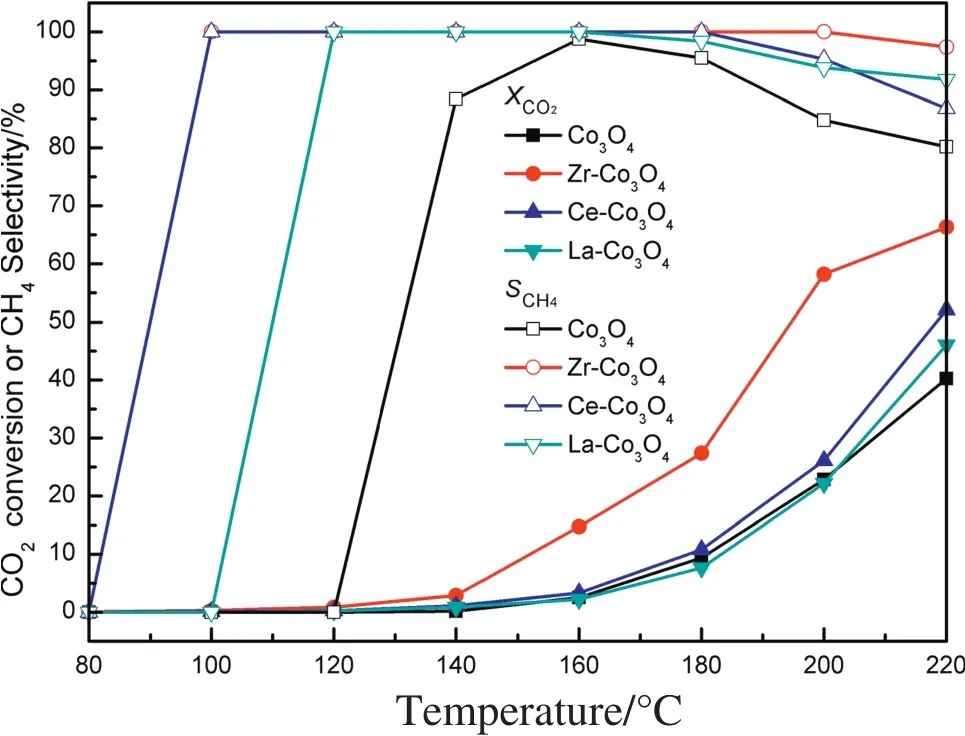
Fig.7.Effects of temperature on catalytic CO2 hydrogenation to methane for Co3O4,La-Co3O4,Ce-Co3O4,Zr-Co3O4.Reaction conditions:T=80–220 °C,P=0.5 MPa,GHSV=18,000 ml··h-1,and V(H2)/V(CO2)=4.
The higher catalytic activity of the Zr-Co3O4than that of La-Co3O4and Ce-Co3O4indicated that Zr plays an important role on improving the CH4synthesis.From a combination of N2adsorption/desorption results,the specific surface area was found to increase when the Zr,Ce,and La was added,and the Zr-Co3O4had a greater specific surface area of71 m2·g-1than thatofthe Co3O4,La-Co3O4,and Ce-Co3O4.Therefore,the increase in the specific surface area by the addition of Zr might be partially responsible forthe greatestimproved effecton the catalytic hydrogenation process.Combined with the XPS and H2-TPR results,modifying the Co3O4with Zr decreased the Co outer-shell electron density and changed the reduction degree of Co3O4by an interaction between Co3O4and ZrO2,and it seems more favorable for CO2methanation when Co3O4were not completely reduced,which would probably increase the CO2conversion and CH4selectivity.Furthermore,from the results of CO2-TPD and pyridine FT-IR spectra,the surface basicity of the catalysts were altered after the addition of Zr by increasing the intensity of the weak and medium basic sites,and the amount of Lewis acids along with the occurrence of Brønsted acid were also enhanced,which would facilitate the activation of CO2under 200°C during the process of CO2hydrogenation,resulting in better catalytic activity.
Moreover,the stabilities of Co3O4and Zr,Ce,or La modi fied Co3O4were carried out on stream for 20 h via CO2methanation at 200°C and 0.5 MPa,with a GHSV=18,000 ml··h-1and V(H2)/V(CO2)=4,as shown in Fig.8.The results showed that the Co3O4catalyst suffered from a large activity loss in 10 h,CO2conversion decreased from 22.5%to 11.4%.In contrast,the Zr-,Ce-,and La-Co3O4catalysts exhibited good catalytic stability,with CO2conversion decreasing from 57.9%,26.4%,and 23.2%to 52.9%,20.6%,and 15.7%in 20 h,indicating the stability of the Co3O4catalyst was improved by modi fied by Zr,Ce,or La.Among them,the Zr-Co3O4exhibited the superior stability.
The higher catalytic activity of the Zr-Co3O4than that of La-Co3O4and Ce-Co3O4indicated that Zr plays an important role on the CH4synthesis.From a combination ofN2adsorption/desorption results,the specific surface area was found increasing when the Zr,Ce,and La was added,and the Zr-Co3O4had a higher specific surface area of 71 m2·g-1than that of other samples.Therefore,the increase in the specific surface area by the addition of Zr might be partially responsible for the improved effect on the catalytic hydrogenation.Combined with the XPS and H2-TPR results,Zr-modi fied on Co3O4decreased the Co outer-shell electron density and changed the reducibility of Co3O4by a stronger interaction between Co3O4and ZrO2,which was favorable for CO2methanation when Co3O4were not completely reduced,and would probably increase the CO2conversion and CH4selectivity.Furthermore,from the CO2-TPD results,the intensity of the weak and medium basic sites was increased after the Zr-addition,which would facilitate the CO2activation during the CO2hydrogenation,resulting in better catalytic activity.
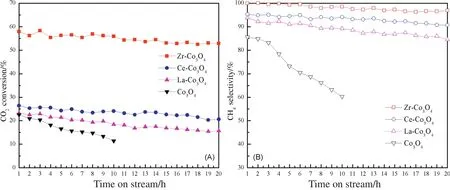
Fig.8.Effects of time on stream on CO2 conversion(A)and CH4 selectivity(B)for the Co3O4,La-Co3O4,Ce-Co3O4,and Zr-Co3O4 catalysts.
4.Conclusions
The Zr-,Ce-,and La-Co3O4catalysts were prepared and applied to the CO2methanation.The results indicated that adding Zr,Ce,or La to the Co3O4catalyst decreased the crystallite sizes of Co,the outer-shell electron density of Co3+,and increased the surface area.The H2-TPR results showed that the reducibility of Co3O4catalyst was significantly changed by adding Zr due to the interaction between Co3O4and ZrO2,which probably provided more active sites(Co2+/Co)for CO2methanation when the Co3O4were notreduced completely.Moreover,the introduction of Zr to the Co3O4catalyst increased the basic intensity of the weak and medium basic sites,as well as the amount of Lewis,and Brønsted acid sites were also found on the Zr-Co3O4catalyst surface,while the Ce and La had little promotion effect on the basic intensity,which predicted that more CO2molecules would activate on the Zr-Co3O4,resulting to higher catalytic activity for CO2methanation.When using the optimalZr-Co3O4with 2.0-wt%ZrO2as the catalyst,and reacted at 200 °C and 0.5 MPa with a GHSV of 18,000 mlh-1,the CO2conversion and CH4selectivity was 58.2%and 100%,respectively.
References
[1]W.Wang,S.Wang,X.Ma,J.Gong,Recent advances in catalytic hydrogenation of carbon dioxide,Chem.Soc.Rev.40(2011)3703–3727.
[2]T.Cantat,L.-N.He,Innovative methods in CO2conversion:a breath of fresh air?Curr.Opin.Green Sustain.Chem.3(2017)iii–iv.
[3]X.D.Xu,J.A.Moulijn,Mitigation of CO2by chemical conversion:plausible chemical reactions and promising products,Energy Fuel 10(1996)305–325.
[4]Q.-W.Song,Z.-H.Zhou,L.-N.He,Ef ficient,selective and sustainable catalysis of carbon dioxide,Green Chem.19(2017)3707–3728.
[5]I.Graca,L.V.Gonzalez,M.C.Bacariza,A.Fernandes,C.Henriques,J.M.Lopes,M.F.Ribeiro,CO2hydrogenation into CH4on NiHNaUSY zeolites,Appl.Catal.B 147(2014)101–110.
[6]W.Zhen,B.Li,G.Lu,J.Ma,Enhancing catalytic activity and stability for CO2methanation on Ni@MOF-5 via control of active species dispersion,Chem.Commun.51(2015)1728–1731.
[7]J.Xu,Q.Lin,X.Su,H.Duan,H.Geng,Y.Huang,CO2methanation over TiO2–Al2O3binary oxides supported Ru catalysts,Chin.J.Chem.Eng.24(2016)140–145.
[8]H.L.Song,J.Yang,J.Zhao,L.J.Chou,Methanation of carbon dioxide over a highly dispersed Ni/La2O3catalyst,Chin.J.Catal.31(2010)21–23.
[9]F.Ocampo,B.Louis,L.Kiwi-Minsker,A.-C.Roger,Effect of Ce/Zr composition and noble metal promotion on nickel based CexZr1-xO2catalysts for carbon dioxide methanation,Appl.Catal.A 392(2011)36–44.
[10]H.C.Lee,K.W.Siew,M.R.Khan,S.Y.Chin,J.Gimbun,C.K.Cheng,Catalytic performance of cement clinker supported nickel catalyst in glycerol dry reforming,J.Energy Chem.23(2014)645–656.
[11]H.Zhu,R.Razzaq,L.Jiang,C.Li,Low-temperature methanation of CO in coke oven gas using single nanosized Co3O4catalysts,Catal.Commun.23(2012)43–47.
[12]P.Munnik,P.E.de Jongh,K.P.de Jong,Control and impact of the nanoscale distribution of supported cobalt particles used in Fischer-Tropsch catalysis,J.Am.Chem.Soc.136(2014)7333–7340.
[13]G.Zhou,T.Wu,H.Xie,X.Zheng,Effects ofstructure on the carbon dioxide methanation performance of Co-based catalysts,Int.J.Hydrog.Energy 38(2013)10012–10018.
[14]G.Zhou,T.Wu,H.Zhang,H.Xie,Y.Feng,Carbon dioxide Methanation on ordered mesoporous Co/KIT-6 catalyst,Chem.Eng.Commun.201(2014)233–240.
[15]A.Y.Khodakov,W.Chu,P.Fongarland,Advances in the development of novel cobalt Fischer-Tropsch catalysts for synthesis of long-chain hydrocarbons and clean fuels,Chem.Rev.107(2007)1692–1744.
[16]S.L.Soled,E.Iglesia,R.A.Fiato,J.E.Baumgartner,H.Vroman,S.Miseo,Control of metal dispersion and structure by changes in the solid-state chemistry of supported cobalt Fischer-Tropsch catalysts,Top.Catal.26(2003)101–109.
[17]Y.Zhu,S.Zhang,Y.Ye,X.Zhang,L.Wang,W.Zhu,F.Cheng,F.Tao,Catalytic conversion of carbon dioxide to methane on ruthenium–cobalt bimetallic nanocatalysts and correlation between surface chemistry of catalysts under reaction conditions and catalytic performances,ACS Catal.2(2012)2403–2408.
[18]X.Zhou,T.Su,Y.Jiang,Z.Qin,H.Ji,Z.Guo,CuO-Fe2O3-CeO2/HZSM-5 bifunctional catalyst hydrogenated CO2for enhanced dimethyl ether synthesis,Chem.Eng.Sci.153(2016)10–20.
[19]Z.Z.Qin,X.H.Zhou,T.M.Su,Y.X.Jiang,H.B.Ji,Hydrogenation of CO2to dimethyl ether on la-,Ce-modi fied Cu-Fe/HZSM-5 catalysts,Catal.Commun.75(2016)78–82.
[20]R.-w.Liu,Z.-z.Qin,H.-b.Ji,T.-m.Su,Synthesis of dimethyl ether from CO2and H2using a Cu–Fe–Zr/HZSM-5 catalyst system,Ind.Eng.Chem.Res.52(2013)16648–16655.
[21]M.C.Bacariza,I.Graça,S.S.Bebiano,J.M.Lopes,C.Henriques,Magnesiumas promoter of CO2Methanation on Ni-based USY zeolites,Energy Fuel 31(2017)9776–9789.
[22]K.Ray,G.Deo,A potential descriptor for the CO2hydrogenation to CH4over Al2O3supported Ni and Ni-based alloy catalysts,Appl.Catal.B 218(2017)525–537.
[23]C.E.Daza,O.A.Gamba,Y.Hernandez,M.A.Centeno,F.Mondragon,S.Moreno,R.Molina,High-stable mesoporous Ni-Ce/clay catalysts for syngas production,Catal.Lett.141(2011)1037–1046.
[24]W.Ahmad,M.N.Younis,R.Shawabkeh,S.Ahmed,Synthesis of lanthanide series(La,Ce,Pr,Eu&Gd)promoted Ni/γ-Al2O3catalysts for methanation of CO2at low temperature under atmospheric pressure,Catal.Commun.100(2017)121–126.
[25]S.Modak,M.Ammar,F.Mazaleyrat,S.Das,P.K.Chakrabarti,XRD,HRTEM and magnetic properties of mixed spinel nanocrystalline Ni-Zn-Cu-ferrite,J.Alloys Compd.473(2009)15–19.
[26]M.Bahmani,B.Vasheghani Farahani,S.Sahebdelfar,Preparation of high performance nano-sized Cu/ZnO/Al2O3methanol synthesis catalyst via aluminum hydrous oxide sol,Appl.Catal.A 520(2016)178–187.
[27]Y.Wang,R.Wu,Y.Zhao,Effect of ZrO2promoter on structure and catalytic activity of the Ni/SiO2catalyst for CO methanation in hydrogen-rich gases,Catal.Today 158(2010)470–474.
[28]L.Samiee,F.Shoghi,A.Vinu,Fabrication and electrocatalytic application of functionalized nanoporous carbon material with different transition metal oxides,Appl.Surf.Sci.265(2013)214–221.
[29]B.A.Sexton,A.E.Hughes,T.W.Turney,An XPS and TPR study of the reduction of promoted cobalt-kieselguhr Fischer-Tropsch catalysts,J.Catal.97(1986)390–406.
[30]J.Li,N.J.Coville,Effect of boron on the sulfur poisoning of Co/TiO2Fischer–Tropsch catalysts,Appl.Catal.A 208(2001)177–184.
[31]A.Feller,M.Claeys,E.van Steen,Cobalt cluster effects in zirconium promoted Co/SiO2Fischer-Tropsch catalysts,J.Catal.185(1999)120–130.
[32]C.I.Ahn,Y.J.Lee,S.H.Um,J.W.Bae,Ordered mesoporous CoMOx(M=Al or Zr)mixed oxides for Fischer-Tropsch synthesis,Chem.Commun.52(2016)4820–4823.
[33]S.Rojanapipatkul,B.Jongsomjit,Synthesis of cobalt on cobalt-aluminate via solvothermal method and its catalytic properties for carbon monoxide hydrogenation,Catal.Commun.10(2008)232–236.
[34]J.-Y.Luo,M.Meng,X.Li,X.-G.Li,Y.-Q.Zha,T.-D.Hu,Y.-N.Xie,J.Zhang,Mesoporous Co3O4–CeO2and Pd/Co3O4–CeO2catalysts:synthesis,characterization and mechanistic study of their catalytic properties for low-temperature CO oxidation,J.Catal.254(2008)310–324.
[35]S.D.Jones,L.M.Neal,M.L.Everett,G.B.Ho flund,H.E.Hagelin-Weaver,Characterization of ZrO2-promoted Cu/ZnO/nano-Al2O3methanol steam reforming catalysts,Appl.Surf.Sci.256(2010)7345–7353.
[36]P.Gao,F.Li,N.Zhao,F.K.Xiao,W.Wei,L.S.Zhong,Y.H.Sun,In fluence of modi fier(Mn,La,Ce,Zr and Y)on the performance of Cu/Zn/Al catalysts via hydrotalcite-like precursors for CO2hydrogenation to methanol,Appl.Catal.A 468(2013)442–452.
[37]L.T.Jia,K.G.Fang,J.G.Chen,Y.H.Sun,Cobalt loss from Co-ZrO2catalyst for Fischer-Tropsch synthesis in continuously stirred tank reactor,React.Kinet.Catal.Lett.93(2008)351–358.
[38]T.Nowitzki,A.F.Carlsson,O.Martyanov,M.Naschitzki,V.Zielasek,T.Risse,M.Schmal,H.J.Freund,M.Bäumer,Oxidation of alumina-supported Co and Co-Pd model catalysts for the Fischer-Tropsch reaction,J.Phys.Chem.C 111(2007)8566–8572.
[39]R.Razzaq,C.Li,M.Usman,K.Suzuki,S.Zhang,A highly active and stable Co4N/γ-Al2O3catalyst for CO and CO2methanation to produce synthetic natural gas(SNG),Chem.Eng.J.262(2015)1090–1098.
[40]G.Busca,Spectroscopic characterization of the acid properties of metal oxide catalysts,Catal.Today 41(1998)191–206.
[41]F.Benaliouche,Y.Boucheffa,P.Ayrault,S.Mignard,P.Magnoux,NH3-TPD and FTIR spectroscopy of pyridine adsorption studies for characterization of Ag-and Cu-exchanged X zeolites,Microporous Mesoporous Mater.111(2008)80–88.
[42]G.R.Johnson,A.T.Bell,Effects of Lewis acidity of metal oxide promoters on the activity and selectivity of Co-based Fischer–Tropsch synthesis catalysts,J.Catal.338(2016)250–264.
[43]X.Tang,J.Li,L.Sun,J.Hao,Origination of N2O from NO reduction by NH3over β-MnO2and α-Mn2O3,Appl.Catal.B 99(2010)156–162.
[44]R.W.Stevens Jr.,S.S.C.Chuang,B.H.Davis,In situ infrared study ofpyridine adsorption/desorption dynamics over sulfated zirconia and Pt-promoted sulfated zirconia,Appl.Catal.A 252(2003)57–74.
猜你喜欢
祖国(2024年1期)2024-01-23 11:08:08
大飞机(2021年4期)2021-07-19 04:41:16
军事文摘(2020年15期)2020-08-15 08:40:02
经济技术协作信息(2018年28期)2018-11-22 05:27:06
小学生学习指导(小军迷联盟)(2018年10期)2018-10-12 01:13:40
人民交通(2018年16期)2018-03-27 01:10:28
 Chinese Journal of Chemical Engineering2018年4期
Chinese Journal of Chemical Engineering2018年4期
- Chinese Journal of Chemical Engineering的其它文章
- An innovative design of septic tank for wastewater treatment and its performance evaluation:An applicable model for developing countries
- Oil field produced water treatment in internal-loop airlift reactor using electrocoagulation/ flotation technique
- From pollutant to solution of wastewater pollution:Synthesis of activated carbon from textile sludge for dye adsorption
- 17α-Ethinylestradiol removal from water by magnetic ion exchange resin☆
- Transesteri fication of sun flower oil in microchannels with circular obstructions
- The extraction of potassium from K-feldspar ore by low temperature molten salt method☆