
极低密度脂蛋白受体在缺血缺氧引起的心肌损伤中的作用研究
2018-05-24 09:50刘志华马晨光
集成技术 2018年3期
刘志华 马晨光
1(中国科学院深圳先进技术研究院 深圳 518055)
2(北京予求医学研究院 北京 100022)
1 引 言
随着人口老龄化的加剧,心血管疾病已经成为威胁人类生命的“头号杀手”。缺血性心肌病亦即冠心病,是一种常见的心血管疾病,是由于冠状动脉粥样硬化引起管腔狭窄,因此造成心肌供血供氧障碍而引起的心脏病。在临床上,缺血性心肌病特别是急性期,病人的死亡率及存活病人心力衰竭的发病率仍然居高不下。缺血性心肌病严重危害着患者的身体健康,对患者乃至其家庭和社会的伤害很大。
一切组织、器官正常的机能、代谢和形态结构的维持,必须有充分的、与代谢相适应的血液灌注。如果血液灌流量绝对或相对不足,均可引起相应组织、器官不同程度的机能、代谢紊乱和结构损害,即缺血性损伤[1]。不同组织、器官对缺血的耐受性不同,甚至有相当大的差异,但这种变化如果发生在重要器官(如心脏),将会给机体带来严重影响。心肌缺血性损伤(Myocardial Ischemic Injury)是由于多种原因引起的冠状动脉管腔狭窄或阻塞而导致,以冠状动脉粥样硬化最常见[2]。
缺血导致心肌发生细胞水平的损伤及器官水平的功能障碍。目前国内外的研究表明,缺血引起心肌细胞氧化应激,表现在细胞内活性氧(ROS)增多、线粒体呼吸链受损[3,4];同时也引起心肌细胞内质网应激出现[5,6],导致细胞凋亡[7-9]及自噬[10,11],严重时引起细胞坏死,即心肌梗死。缺血引起心肌组织严重的炎症反应,表现在炎细胞浸润,包括巨噬细胞、中性粒细胞及 T 细胞等,同时炎细胞及心肌细胞释放多种炎症因子,包括 IL-1β、IL-6、IL-10、TNF-α、MCP-1等[12,13]。这些病理学的改变使心肌细胞出现可逆或不可逆的损伤,对心功能造成极为不利的影响。
一直以来,缺血性心肌损伤的分子病理机制备受国际关注,国内外研究已经发现了许多分子和信号通路参与了其中的发生、发展[14-19]。相关的机理涉及多种受体分子(如腺苷受体、α1受体、AngⅡ 受体和 M2受体等)[20],各种跨膜信号转导机制(如受体-抑制性 G 蛋白-腺苷环化酶系统、受体-磷脂酶 C-蛋白激酶 C 系统、NO-cGMP 信号转导系统)[21,22],多种信号通路(如MAPKs、AKT、NF-κB)[23,24],同时还包括各种内源性保护蛋白(如热休克蛋白、金属硫蛋白、氧自由基清除系统)[25]。然而,缺血性心肌损伤是个复杂的病理过程,这些被发现的分子并不能彻底地诠释它的病理表现,其中的许多机制还有待探讨。极低密度脂蛋白(Very Low Density Lipoprotein,VLDL)受体是低密度脂蛋白受体超基因家族的一员,能以高亲和力结合含有 apoE的脂蛋白,是机体细胞摄取血液中富含甘油三脂的 VLDL 的主要受体,并与低密度脂蛋白受体在结构上极其相似。这两种受体均含有 5个功能域:(1)具有含半胱氨酸的数个重复序列;(2)EFG 同源域;(3)丝氨酸和苏氨酸富集区,此区域是受体细胞外紧接胞膜的部分,被认为是以 O-O 碳水化合物簇的部位;(4)跨膜部分;(5)胞内域,含有一个包被陷窝的靶信号[26]。1992年,Lijima 等[27]成功地克隆了兔 VLDL 受体的 cDNA,阐明了其氨基酸序列及功能域,并基于对家兔内各组织 VLDL 受体结构及功能的研究成果,将 VLDL 受体分为两型:Ⅰ 型为含有O-Link 糖链结合域,主要分布于骨骼肌、心肌、脂肪等组织中;Ⅱ 型则不含有糖链结合域,主要分布于脂肪、脾脏、肾脏等非肌组织中。Ⅰ 型受体比 Ⅱ 型受体难降解,结合 VLDL 的能力也更强。因此,推测 Ⅰ 型 VLDL 受体在摄取甘油三酯中发挥了更大作用。
现阶段存在的问题是:国内外研究并未充分阐明 VLDL 受体的生物学功能。多数研究注重于VLDL 受体与能量代谢、脂类代谢的关系,即便如此,VLDL 受体在能量代谢中的分子作用也未明确定义。关于 VLDL 受体的其他生物学功能研究很有限,而这些有限的研究却表明 VLDL 受体不只是参与脂质代谢那么简单,它在细胞增殖、细胞凋亡、血管新生,甚至炎症反应[28-31]中发挥了调控作用。同时,关于 VLDL 受体发挥生物学功能的分子机理也有待明确。有研究表明,它对AKT 信号通路具有抑制作用[30]。VLDL 受体在心血管疾病中的作用更是少有研究,而且多局限在内皮细胞和血管平滑肌细胞[29,30]。但这些不多的研究却提示 VLDL 受体与心血管疾病的发生、发展密切相关[31]。关于 VLDL 受体在心肌病理损伤中的作用,目前仅检索到 1篇发表在 Journal of Clinical Investigation 期刊上的成果。该文对VLDL 受体在心肌凋亡中的作用进行研究,但关于心肌损伤的其他表型及其中的分子机理,文中并未深入探讨[32]。本文结合生物信息学和生物医学实验的方法和技术,通过开展实验验证,旨在研究并阐明 VLDL 受体对小鼠缺血性心肌损伤的影响及其中的分子机制。
2 实验方案
2.1 实验动物分组与模型的建立
实验动物包括:野生型(Wild Type,WT)小鼠、极低密度脂蛋白受体基因敲除(VLDLR-KO)小鼠及心脏特异性转基因(VLDLR-TG)小鼠。选择体重在 24~26g、8周龄的雄性小鼠为实验对象,建立缺血性心肌损伤小鼠模型,进行观察和研究。
具体实验分为假手术(Sham)组和缺血(Ischemia)手术组两组。其中,两个实验组均包括 WT 小鼠、VLDLR-KO 小鼠及 VLDLR-TG 小鼠各 50只。
对于缺血(Ischemia)手术组,首先,小鼠戊巴比妥钠腹腔注射麻醉,左侧胸部备皮,消毒手术区域,插管连接小动物呼吸机,沿第 3~4肋间水平切开皮肤,依次分离肌肉及软组织,暴露心脏,于肺动脉圆锥与左心耳之间距主动脉根部 1mm 处结扎前降支 30min,观察缝线下方区域变白,局部心肌运动减弱。然后,逐层缝合胸壁,待恢复自主呼吸后拔出通气导管。最后,待动物清醒后送回饲养室,自由进食和饮水。而假手术(Sham)组只在前降支处穿过一条短丝线,不作结扎,其他步骤同缺血(Ischemia)手术组(即模型组)。
无菌操作取出 1~3天乳鼠心脏,D-Hanks液冲洗,剪碎心室,用 0.06% II 型胶原酶和 25%胰酶分次消化,离心收集细胞后用含 15% 胎牛血清的 DMEM/F12培养液悬浮分散细胞,接种于 100mm 培养皿中,37℃、5% CO2差时贴壁 1.5h。上清接种到明胶包被的 6孔板中,以α-actinin 染色鉴定心肌细胞纯度。
取原代培养至第 4天的细胞,用磷酸盐缓冲液(PBS)洗涤细胞 2~3次,然后换成无血清的培养基,并放至 37℃、5% CO2、95% N2密闭的缺氧装置中培养 16h,以此体外模拟细胞缺血缺氧状态。
2.2 缺血缺氧引起心肌损伤反应指标的检测
基于已经建成的缺血性心肌损伤小鼠模型和体外细胞缺血缺氧模型,对分组的动物进行相关指标的检测,主要包括炎症反应、细胞坏死、凋亡和自噬的检测。相比于传统的检测指标,我们重点关注缺血缺氧引起的心肌细胞的死亡途径,包括细胞凋亡、细胞坏死和自噬。具体检测流程如下:
采用氧化应激指标检测试剂盒进行相应检测。具体地,用 Fe3+还原法测定血浆 T-AOC,采用 Fenton 反应及 Gress 显色原理测定细胞内活性氧(ROS),黄嘌呤氧化酶法测定超氧化物歧化酶(SOD),硫代巴比妥酸法(TBA)测定丙二醛(MDA)含量,硝酸还原酶法测定一氧化氮(NO)。
应用实时聚合酶链反应(Real-time PCR)与蛋白质印迹(Western blot)检测组织及细胞中内质网应激标志分子 GRP78、Chop、Atf6、Bip、XBP-1的表达水平;同时应用 Western blot 检测组织及细胞中 PERK 及 c-jun 的磷酸化水平。
2.2.1 炎症反应的检测
(1)心肌组织炎症反应的检测
①炎性细胞的浸润:检测缺血区巨噬细胞、中性粒细胞及 T 细胞的数量。各组心肌组织的冰冻切片用 Mac3、CD45、CD4、CD8及 7/4抗体进行免疫组化染色,计数每组染色阳性细胞数。
②炎症因子检测:目前,心肌细胞代表性炎症因子有 IL-1β、IL-6、IL-10、TNF-α、MCP-1。通过提取组织总 RNA 和蛋白质,应用 Realtime PCR 与 Western blot 可测定这些炎症因子的表达。
(2)心肌细胞炎症反应检测
采用酶联免疫吸附测定(ELISA)和 Real-time PCR 检测培养上清及心肌细胞炎症因子 IL-1β、IL-6、IL-10、TNF-α、MCP-1的表达。
2.2.2 细胞坏死、凋亡和自噬的检测
采用 Roche 公司的 In situ Cell Death Detection(POD)试剂盒检测各组凋亡细胞,同时采用 Western bolt 检测凋亡相关分子 Bcl2、Bax、Bad、Bcl-xl、cIAP、FLIP 及 caspase8/cleaved caspase8、caspase9/cleaved caspase9、caspase3/cleaved caspase3等蛋白表达水平;采用PI(Propidium Iodide)染色检测细胞的坏死;电镜下观察细胞内的自噬体,同时用 Western bolt 检测 LC3Ⅱ、LC3Ⅰ 的表达水平。
3 结果与分析
3.1 VLDL 受体对缺血性心肌损伤的影响途径研究
本文通过在体实验结合离体实验研究 VLDL受体对缺血性心肌损伤的影响。
在体实验中,通过采用 C57品系的野生型小鼠、VLDLR 基因敲除小鼠及心脏特异性 VLDLR转基因小鼠(均以 C57为背景建立),建立左冠状动脉结扎诱导的小鼠心肌缺血动物模型,研究假手术组(Sham)与手术组(Ischemia)之间,以及野生型组与基因敲除组及转基因组之间在心肌损伤方面的差异。
离体实验中,分离以上 3种乳鼠的心肌细胞,通过正常培养或缺血缺氧培养,以比较正常培养组与缺血缺氧培养组之间,以及野生型组与基因敲除组及转基因组的心肌细胞之间在心肌损伤方面的差异。
具体指标有:
(1)氧化应激:测定血浆总抗氧化能力(T-AOC),测定组织及细胞内活性氧(ROS)、超氧化物歧化酶(SOD)、丙二醛(MDA)及一氧化氮(NO)含量。
(2)内质网应激:检测组织及细胞中 PERK、Bip、XBP-1、Chop、Atf6及 c-jun 等的表达水平。
(3)炎症:检测心肌组织中炎症细胞的浸润,包括巨噬细胞、中性粒细胞及 T 细胞的浸润;检测组织及细胞中 IL-6、IL-10、IL-1β、TNF-α、MCP-1的表达。
(4)细胞坏死:测定血浆谷草转氨酶(GOT)、肌酸激酶(CK)、乳酸脱氢酶(LDH)的水平;检测心肌组织的坏死面积及细胞实验中细胞坏死的程度。
(5)细胞凋亡:检测在体及离体实验中凋亡细胞的数量;检测组织及细胞中凋亡相关分子 Bcl2、Bax、Bad、Bcl-xl、cIAP、FLIP及 caspase8/cleaved caspase8、caspase9/cleaved caspase9、caspase3/cleaved caspase3等的表达。
(6)细胞自噬:检测自噬体的形成及自噬相关分子 LC3Ⅱ/LC3Ⅰ 的比值。
具体研究内容如下:
(1) 缺血缺氧诱导心肌细胞 VLDL 受体的表达
乳鼠心肌细胞缺血缺氧培养 16h 后,采用Western blot 检测 VLDL 受体蛋白水平,结果如图1所示。图1结果表明,缺血缺氧能诱导心肌细胞 VLDL 受体的表达。
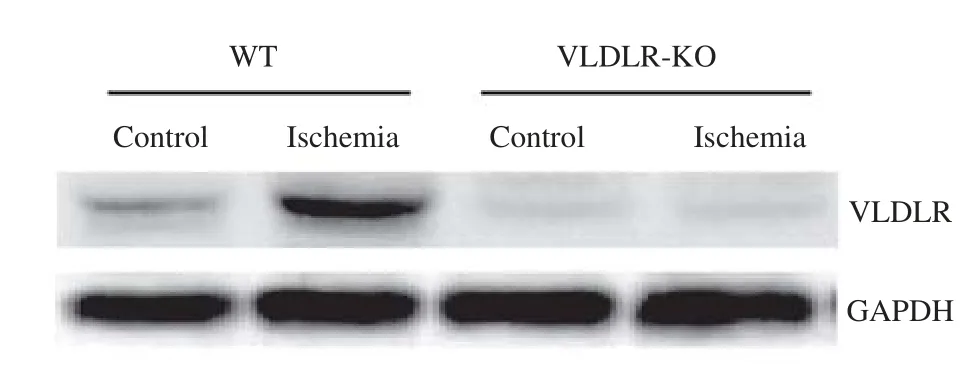
图1 缺血缺氧引起心肌细胞 VLDL 受体(VLDLR)表达增强Fig.1Increased expression of VLDL receptor(VLDLR) in myocardial cells induced by ischemia and hypoxia
(2) VLDL 受体缺失保护缺血缺氧引起的心肌坏死和凋亡
在小鼠接受心肌 Ischemia 手术组中,术后3天分别采用苏木精-伊红(Hematoxylin-Eosin,HE)染色法检测组织坏死程度、 TUNEL 染色检测细胞凋亡水平,结果如图2所示。图2结果表明,VLDL 受体缺失保护缺血缺氧引起的心肌坏死和凋亡。
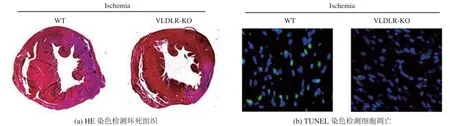
图2 VLDL 受体缺失保护缺血缺氧引起的心肌坏死和凋亡Fig.2VLDL receptor deletion protects against myocardial necrosis and apoptosis induced by ischemia and hypoxia
(3) VLDL 受体缺失抑制缺血缺氧引起的心肌组织 IL-1β 和 TNF-α 的表达
在小鼠接受心肌 Ischemia 手术组中,术后 3天采用免疫组化染色检测 CD3和 CD45阳性细胞的组织侵润;乳鼠心肌细胞缺血缺氧培养 16h 后用 ELISA 检测培养上清液中 IL-1β 和 TNF-α 的水平,结果如图3所示。结果表明,VLDL 受体缺失抑制缺血缺氧引起的心肌组织 IL-1β 和 TNF-α的表达。

图3 VLDL 受体缺失抑制缺血缺氧引起的心肌组织炎症反应Fig.3VLDL receptor deletion inhibits infl ammation of myocardial tissue induced by ischemia and hypoxia
(4) VLDL 受体缺失抑制缺血缺氧引起的p-AKT 水平下调
乳鼠心肌细胞缺血缺氧培养 16h 后用Western blot 检测 p-AKT 蛋白水平,结果如图4所示。结果表明,VLDL 受体缺失抑制缺血缺氧引起 p-AKT 水平下调。

图4 VLDL 受体缺失缓解缺血缺氧引起的 p-AKT 水平下调Fig.4VLDL receptor deletion reduces p-AKT levels induced by ischemia and hypoxia
通过以上在体实验结合离体实验的检测发现:缺血缺氧可以诱导心肌细胞 VLDL 受体的表达;而 VLDL 受体缺失不但可以保护缺血缺氧引起的心肌坏死和凋亡,还可以抑制缺血缺氧引起的心肌组织 IL-1β 和 TNF-α 的表达,及可以抑制缺血缺氧引起的 p-AKT 水平下调。
3.2 VLDL 受体影响缺血性心肌损伤的分子机制的讨论
为了使本研究具有系统性和全面性,我们首先开展高通量芯片实验,然后进行生物信息学分析,最后对实验结果开展生物医学实验验证。即生物学实验结合生物信息学分析的研究思路,通过构建生物分子网络探讨 VLDL 受体影响缺血性心肌损伤的分子机制,包括关键分子与关键信号通路两个方面。具体如下:
我们对芯片原始数据进行标准化处理,消除背景噪音,去除同一实验的不同芯片之间的实验误差。具体地,采用 RMA 算法进行背景校正,采用 Quantile 算法进行数据标准化,采用 Pmonly 算法进行 PM(Perfect Match)探针校正,采用 Medianpolish 算法进行表达值的评估。数据标准化后采用 Limma 进行差异表达分析,以 P<0.01为筛选条件,筛选差异表达基因。选取 FDR(False Discovery Rate)<0.05的差异表达基因,以蛋白互作数据、转录调控数据、翻译后修饰数据、生物信号通路数据为背景信息,应用Cytoscape 建立以 VLDL 受体为核心分子的生物分子网络。
3.2.1 VLDL 受体发挥生物学功能的关键分子研究
生物信息学技术发现关键分子:本文采用高通量的基因芯片实验,发现 VLDL 受体影响哪些基因的表达,即获得心肌损伤情况下 VLDL受体影响的差异表达基因数据集。采用 Broad Institute of MIT and Harvard 开发的 GSEA 算法进行分析。具体是以 GO(Gene Ontology)、TFT(Transcription Factor Targets)、KEGG、Biocarta 数据库为信息背景,以 nominal P_value<0.05为筛选条件,分别进行生物学过程(Biological Process)、转录因子、信号通路的功能富集分析。
本文发现关键分子的过程包括:(1)对差异表达基因以 GO 数据库和 TFT 数据库为分析背景,进行功能富集分析,发现属于各生物学过程的重要分子及表达调控的转录因子;(2)以蛋白互作数据、转录调控数据、翻译后修饰数据为信息建立分子网络,基于图论原理(Graphic Theory)发现网络的拓扑关系及关键节点分子。分析生物网络的拓扑学参数,包括度(Degree)、聚类系数(Clustering Coefficient)、介数(Betweenness)、紧密度(Closeness)、同配性系数(Assortativity Coefficient)综合评价分子节点在网络中的作用权重,发现 VLDL 受体为网络中发挥生物学功能的关键分子。
另外,本文还通过生物医学实验进一步验证了关键分子:检测缺血缺氧状态下,上述发现的关键分子分别在培养的 C57、VLDLR-KO及 VLDLR-TG 乳鼠心肌细胞中的表达水平,确定 VLDL 受体对上述发现的关键分子是否具有影响;同时,对关键分子也进行基因敲除(如siRNA 转染)或基因过表达(如腺病毒转染)实验,检测关键分子是否影响缺血性心肌损伤的各种病理表型(前部分提到的氧化应激、炎症、凋亡、自噬等);最后,综合结果确定 VLDL 受体→分子→心肌损伤病理表型的因果关联性。
3.2.2 VLDL 受体发挥生物学功能的关键信号通路研究
(1)基于芯片结果发现关键信号通路
基于生物信息学技术发现关键信号通路:以KEGG 和 Biocarta 数据库为分析背景,对心肌损伤情况下 VLDL 受体影响的差异表达基因数据集进行功能富集分析,计算显著被抑制及显著被激活的信号通路。同时,检测缺血缺氧状态下,VLDL 受体是否对上述关键信号通路及其中的分子存在影响;另外对关键信号通路或其中的分子进行抑制或激活,检测关键信号通路是否影响缺血性心肌损伤的病理表型;最后,综合结果分析VLDL 受体→信号通路→心肌损伤病理表型的因果关联性。
(2)研究 VLDL 受体对常见的信号通路的影响
功能模块是生物网络中连接紧密的子网络,其紧密度可以用模块度(Modularity,Q)来衡量,它是该子网络中已有的连接数与子网络中可能的最大连接数的比值。计算公式为:

其中,m 为子网络中已有的连接数;n 为子网络中节点的个数。本文取 Q 值大于 0.6进行功能模块分析。
传统以单个和少数几个基因或蛋白的研究方法有很大的局限性。本文采用生物网络的功能模块计算分析,从整体水平上进行研究,发现了 MAPKs(ERK、p38、JNK)、AKT、NF-κB 信号通路在缺血性心肌损伤的发生、发展中起到重要作用,VLDL 受体抑制 AKT 信号通路,然而关于它对 AKT 及其他信号通路在心肌组织中的影响未见研究。本文检测了缺血缺氧状态下,VLDL 受体是否影响这些信号通路;同时,对MAPKs、AKT、NF-κB 信号通路进行抑制或激活,检测它们是否影响缺血性心肌损伤的病理表型;最后,综合各种结果分析并明确了 VLDL 受体→MAPKs、AKT、NF-κB 信号通路→心肌损伤病理表型的因果关联性。
4 结 论
本文应用 C57BL/6品系的野生型小鼠、VLDL 受体基因敲除小鼠和心脏特异性转基因小鼠,通过在体与离体实验系统研究了 VLDL 受体对小鼠缺血性心肌损伤的影响。另外,检测了缺血缺氧状态下,VLDL 受体对这些信号通路的影响;同时,对 MAPKs、AKT、NF-κB 信号通路进行抑制或激活,观察它们对缺血性心肌损伤的病理表型的影响;最后,综合实验结果分析并明确了 VLDL 受体→MAPKs、AKT、NF-κB 信号通路→心肌损伤病理表型的因果关联性,阐明了VLDL 受体影响缺血性心肌损伤的分子机制。
参 考 文 献
[1]Cheung JY,Bonventre JV,Malis CD,et al.Calcium and ischemic injury [J].The New England Journal of Medicine,1986,314(26): 1670-1676.
[2]Kim ES,Sun JK,Park N,et al.Purpose in life and reduced risk of myocardial infarction among older U.S.adults with coronary heart disease: a twoyear follow-up [J].Journal of Behavioral Medicine,2012,36(2): 124-133.
[3]Dreier JP,Drenckhahn C,Woitzik J,et al.Spreading ischemia after aneurysmal subarachnoid hemorrhage [J].Acta Neurochirurgica Supplement,2013,115: 125-129.
[4]Iadecola C,Ross ME.Molecular pathology of cerebral ischemia: delayed gene expression and strategies for neuroprotection [J].Annals of the New York Academy of Sciences,1997,835: 203-217.
[5]Osada N,Kosuge Y,Ishige K,et al.Characterization of neuronal and astroglial responses to ER stress in the hippocampal CA1area in mice following transient forebrain ischemia [J].Neurochemistry International,2010,57(1): 1-7.
[6]Vekich JA,Belmont PJ,Thuerauf DJ,et al.Protein disulfide isomerase-associated 6is an ATF6-inducible ER stress response protein that protects cardiac myocytes from ischemia/reperfusionmediated cell death [J].Journal of Molecular and Cellular Cardiology,2012,53(2): 259-267.
[7]Fujita K,Yoshimoto N,Kato T,et al.Lycopene inhibits ischemia/reperfusion-induced neuronal apoptosis in gerbil hippocampal tissue [J].Neurochemical Research,2013,38(3): 461-469.
[8]Cheon SY,Cho KJ,Lee JE,et al.Cerebroprotective effects of red ginseng extract pretreatment against ischemia-induced oxidative stress and apoptosis[J].International Journal of Neuroscience,2013,123(4): 269-277.
[9]Ding ZM,Wu B,Zhang WQ,et al.Neuroprotective effects of ischemic preconditioning and postconditioning on global brain ischemia in rats through the same effect on inhibition of apoptosis[J].International Journal of Molecular Sciences,2012,13(5): 6089-6101.
[10]Xu F,Li J,Ni W,et al.Peroxisome proliferatoractivated receptor-gamma agonist 15d-prostaglandin J2mediates neuronal autophagy after cerebral ischemia-reperfusion injury [J].PLoS One,2013,8(1): e55080.
[11]Ma X,Liu H,Foyil SR,et al.Autophagy is impaired in cardiac ischemia-reperfusion injury [J].Autophagy,2012,8(9): 1394-1396.
[12]Guzzardi MA,Iozzo P.Fatty heart,cardiac damage,and inflammation [J].The Review of Diabetic Studies,2011,8(3): 403-417.
[13]Gruson D,Ahn SA,Rousseau MF.Biomarkers of infl ammation and cardiac remodeling: the quest of relevant companions for the risk stratification of heart failure patients is still ongoing [J].Biochemia Medica,2011,21(3): 254-263.
[14]刘志华,马晨光.缺血性心肌病不同阶段基因表达的改变模式研究 [J].集成技术,20176(5): 69-75.
[15]Li Y,Jiang Q,Ding Z,et al.Identification of a common different gene expression signature in ischemic cardiomyopathy [J].Genes,2018,9(1):56.
[16]Liu ZH,Yang D,Xie P,et al.MiR-106b and MiR-15b modulate apoptosis and angiogenesis in myocardial infarction [J].Cellular Physiology and Biochemistry,2012,29(5-6): 851-862.
[17]Ren GM,Liu ZH.NetCAD: a network analysis tool for coronary artery disease associated PPI network[J].Bioinformatics,2013,29(2): 279-280.
[18]Cheng JK,Cao FL,Liu ZH.AGP: a multimethods web server for alignment-free genome phylogeny[J].Molecular Biology and Evolution,2013,30(5):1032-1037.
[19]Yang D,Xie P,Liu ZH.Ischemia/reperfusioninduced MKP-3impairs endothelial NO formation via inactivation of ERK1/2pathway [J].PLoS One,2012,7(7): e42076.
[20]Izmirly PM,Buyon JP,Saxena A.Neonatal lupus: advances in understanding pathogenesis and identifying treatments of cardiac disease [J].Current Opinion in Rheumatology,2012,24(5):466-472.
[21]Schluter KD,Wenzel S.Angiotensin II: a hormone involved in and contributing to pro-hypertrophic cardiac networks and target of anti-hypertrophic cross-talks [J].Pharmacology & Therapeutics,2008,119(3): 311-325.
[22]Booz GW.Putting the brakes on cardiac hypertrophy: exploiting the NO-cGMP counterregulatory system [J].Hypertension,2005,45(3):341-346.
[23]Burgoyne JR,Mongue-Din H,Eaton P,et al.Redox signaling in cardiac physiology and pathology [J].Circulation Research,2012,111(8): 1091-1106.
[24]Stahl GL,Shernan SK,Smith PK,et al.Complement activation and cardiac surgery: a novel target for improving outcomes [J].Anesthesia &Analgesia,2012,115(4): 759-771.
[25]Willis MS,Patterson C.Hold me tight: role of the heat shock protein family of chaperones in cardiac disease [J].Circulation,2010,122(17): 1740-1751.[26]Takahashi S,Sakai J,Fujino T,et al.The very low-density lipoprotein(VLDL) receptor:characterization and functions as a peripheral lipoprotein receptor [J].Journal of Atherosclerosis and Thrombosis,2004,11(4): 200-208.
[27]Lijima H,Miyazawa M,Sakai J,et al.Expression and characterization of a very low density lipoprotein receptor variant lacking the O-link sugar region generated by alternative splicing [J].The Journal of Biochemistry,1998,124(4): 747-755.
[28]Liu ZG,Li H,Li YH,et al.Up-regulation of VLDL receptor expression and its signaling pathway induced by VLDL and β-VLDL [J].Journal of Huazhong University of Science and Technology[Medical Sciences],2009,29(1): 1-7.
[29]Jiang A,Hu W,Meng H,et al.Loss of VLDL receptor activates retinal vascular endothelial cells and promotes angiogenesis [J].Investigative Ophthalmology & Visual Science,2009,50(2):844-850.
[30]Oganesian A,Armstrong LC,Migliorini MM,et al.Thrombospondins use the VLDL receptor and a nonapoptotic pathway to inhibit cell division in microvascular endothelial cells [J].Molecular Biology of the Cell,2008,19(2): 563-571.
[31]Perman JC,Boström P,Lindbom M,et al.The VLDL receptor promotes lipotoxicity and increases mortality in mice following an acute myocardial infarction [J].The Journal of Clinical Investigation,2011,121(7): 2625-2640.
[32]Imagawa M,Takahashi S,Zenimaru Y,et al.Comparative reactivity of remnant-like lipoprotein particles(RLP) and low-density lipoprotein(LDL)to LDL receptor and VLDL receptor: effect of a high-dose statin on VLDL receptor expression [J].Clinica Chimica Acta,2012,413(3-4): 441-447.
猜你喜欢
世界科学技术-中医药现代化(2022年2期)2022-05-25
世界科学技术-中医药现代化(2021年7期)2021-11-04
农村青少年科学探究(2020年5期)2020-08-18
中医眼耳鼻喉杂志(2019年3期)2019-04-13
新民周刊(2018年8期)2018-03-02
饮食科学(2017年12期)2018-01-02
中成药(2017年8期)2017-11-22
中国民族医药杂志(2016年5期)2016-05-09
中国病理生理杂志(2015年8期)2015-12-21
药学研究(2015年11期)2015-12-19
