
16S rDNA特异性引物设计优化及其在淞江鲈体表微生物鉴定中的应用
2018-05-15 12:22徐鹏昊罗武松何恩明王焱磊窦同海王金秋
复旦学报(自然科学版) 2018年1期
徐鹏昊,罗武松,何恩明,王焱磊,蔡 枫,张 亮, 窦同海,王金秋,周 雁,
(1.复旦大学 生命科学学院 微生物学与微生物工程系,上海 200438;2.复旦大学 生命科学学院 遗传学研究所 遗传工程国家重点实验室,上海 200438;3.上海人类基因组研究中心 上海市疾病与健康基因组学重点实验室,上海 201203)
PCR引物设计中,引物与模板结合的特异性是首要考虑要素.特异性不足时,引物容易与近似序列发生非特异性结合,产生不纯的扩增产物,在qPCR的时候也会影响定量结果.引物的特异性是保证理想PCR产物的基本条件.
一般而言,较高的引物特异性容易得到单一的PCR产物,但是这并不总是符合实验设计的需求.从属水平上来看,利用16S rDNA分析各种微生物的相对丰度时,设计的引物就需要满足以下两个主要条件: 一是在属内具有较好的覆盖度,能够与属内绝大多数物种的DNA序列结合,防止属内某些种不能扩增出产物而产生误差;二是不与属外的其他物种的DNA结合,防止非特异性扩增的发生.只有同时符合这两个条件,才可以得到较精准的菌群分析结果.同样,在致病菌初期筛查中,也需要控制引物的特异性.同属的微生物种间序列相似度极大[1],也常常具有相似的结构和习性.故在鉴定微生物致病性时,常常以属作为分类单位,并不特指到具体的种.如黄杆菌属(Flavobacterium)、希瓦氏菌属(Shewanella)和假单胞菌属(Pseudomonas)的微生物均会引起淡水鱼类的皮肤溃烂[2-4].因而,在通过PCR方法对这些致病菌进行检测的时候,为了确定这些属的微生物是否存在,也需要属水平上的引物特异性.
现在常用的引物设计软件,如Oligo[5]、Primer Premier和NCBI的Primer-BLAST[6]等,在设计针对属水平的特异性引物时均存在一定局限.其中Oligo和Primer Premier主要针对引物的效率,没有包含模板外特异性检测的模块.Primer-BLAST基于NCBI数据库,设计了引物特异性检测的相关参数,但是不能针对具体环境来设计特异性检测数据库,也不能检测引物在属内的覆盖率.因此,本研究尝试对引物设计进行优化,以满足属水平上引物设计的需求.
引物设计时,选择特异性较好的基因作为模板可以提高引物的特异性,为了了解这样的引物特异性是否满足微生物丰度检测的需要,我们用BLAST结果进行评估.本研究中选取细菌的16S rDNA作为模板,因为其在微生物中体现的高度属间特异性和属内保守性,常作为微生物分类的主要依据[7].由于16S rDNA是原核生物普遍具有的DNA序列,以其为靶基因,可以方便而又准确地检测目标属在样本中是否存在,由此可以对菌群丰度进行分析,或是检测物种是否有被致病微生物感染,产生疾病的风险.我们选取了一些以往实验中使用的16S rDNA特异性引物,验证其在淞江鲈体表微生物菌群分析的效果.发现这些引物都会与其他环境属微生物的DNA发生非特异性结合,或者只能与属内少数几个种匹配,因而不能满足微生物菌群鉴定或是致病微生物检测的需要.因此,本研究的目的,就是针对16S rDNA,在属水平特异性上进行引物设计优化,满足属间特异性和属内覆盖度两方面的需求,以期在微生物菌群分析、致病微生物鉴定等研究中实际应用.
1 设计方法
为了保证引物的属间特异性,同时保证引物在属内的覆盖度,引物经过了两次的筛选,第一次筛选找出属内保守片段,第二次保证引物的特异性,不与其他的DNA发生非特异性扩增.引物设计的流程图见图1.

图1 引物设计流程图Fig.1 Flow diagram of primer design
1.1 确定目标属与环境属
在引物设计中,首先确定检测的目标属.除此之外,为了排除其他DNA发生非特异性扩增的可能,也要确定环境中可能存在的其他属,即环境属.这一步中,为了确定环境属,首先应检索和引用已发表的测序结果或相关菌群分析结果,也可以放宽至所有理论上可能存在的属.一般而言,如果能够通过测序、菌群分析等手段精准地确定环境属,则可以得到更多的备选引物对,从而为兼顾引物的效率提供更多选择.
1.2 下载序列
得到了所有属的列表之后,下载其16S rDNA序列作为引物设计的模板,并转化为FASTA格式.现在常用的数据库,如NCBI(http:∥www.ncbi.nlm.nih.gov/)、RDP[8]等,都提供了多种格式的数据免费下载服务,使得这一步骤较易实现.此外,还需要下载所有环境属的16S rDNA序列,这些序列构成了引物设计软件的原数据.
此外,在确定目的基因时,要尽量保证目的基因或其同源基因在所有属中均存在.因此,优先选择一些功能重要的管家基因,比如说在微生物中,就可以使用16S rDNA作为模板.也可以将目的基因序列在目标属中进行BLAST,并以此检验是否有同源基因存在.同时,尽量保证该基因的属内保守性和属间特异性,具有以上特点的基因可以更好地用于引物设计.
1.3 通用性筛选
通用性筛选,即筛选可以作为引物模板的,在目标属内普遍存在的属内保守片段.这些片段将作为之后引物设计的基础.在这一步中,我们筛选得到的片段数量与目的基因的选择直接相关.16S rDNA属于较为保守的基因片段,可以得到属内保守片段的数量较多,给后续的筛选留下了较为充足的空间.
1.4 特异性筛选
特异性筛选,即确定上一步筛选的片段在属间的特异性.将属内保守片段与所有环境属序列进行比对,选择只在目标属之内出现,与其他环境属的序列之间都有差异的片段,软件中设定的默认最小差异值是属内保守片段与其他序列至少有一个碱基的不同,以排除非特异性扩增.之后,根据产物长度将特异性片段进行配对,得到特异性片段对.
1.5 引物筛选
我们设计了一个简单的引物效率计算模块.通过控制引物之间的二级结构来保证引物的扩增效率.首先,根据引物中ATCG各碱基的比例对引物的tm(退火温度)值进行估算,控制前后引物的tm差值在一定的范围内.之后,对引物序列进行分析,严格控制引物之间的二级结构,包括发夹结构、二聚体等,将这些具有二级结构的引物排除.最后,由于连续相同的碱基可能会对引物的效率产生影响,软件也排除了这些引物.以上几点作为引物筛选的参数,在控制引物效率方面起到了重要作用.
1.6 确定引物对
完成引物效率计算,排除tm差值较大和具有二级结构的引物之后,将特异性片段对的后部分反向互补,就得到了最终引物对.这些引物在只有环境属的情况下,都具有较好的属间特异性和属内覆盖度.有些属由于目的基因特异性不足,可能没有引物对产生.此时取m条特异性片段,m缺省为1000,并取这些片段之后nbp位置的普通片段,将两者进行配对,n默认为最短产物长度值,即200.经过筛选后,得到的引物对前引物具有特异性,后引物的特异性不能保证,因此这样的引物对略逊于前者,为次优引物对.然而,由于前引物的特异性,这样的引物对也可以在扩增中使用.最终确定引物时,可以将模板上不同位置的几对引物,使用BLAST进行检验,选取其中属间特异性好,属内覆盖度高的作为最终引物对.
2 实验验证方法
淞江鲈是中国国家二级保护动物,是一种珍贵的生物资源,以其重要的生态和经济价值位列我国四大名鱼之首[9].养殖淞江鲈的体表常出现皮肤溃烂现象,本实验室对淞江鲈体表菌群进行了测序分析,初步推测了其潜在致病菌[10].为了确定引物设计的特异性和效率,本研究选择淞江鲈作为实验物种,通过定量PCR验证与前期研究结果是否一致.

实验中使用的淞江鲈采集自上海市松江区的上海四鳃鲈水产科技发展有限公司的生产基地,选取了头部严重感染的个体.用2个无菌棉签分别擦拭溃烂部位和周边未感染部位.之后将棉签迅速放入灭菌离心管中,-20℃保存,得到了2个样本(表1).
2.1 16S rDNA库的构建
实验参照文献确定了淞江鲈的3个致病属(Flavobacterium、Pseudomonas和Shewanella)以及一个辅助检测属(Rhodobacter)[10],致病属在致病淞江鲈样本中的含量较高,并会导致辅助检测属的含量有一定降低,因此将其作为实验的目标属.辅助检测属Rhodobacter是淡水环境中的正常菌群[11],同时它的丰度较大[10],便于检测.因而我们将其作为定量PCR的参照.之前的测序结果共发现154个环境属[10],其中在RDP数据库中检索获得142个环境属的16S rDNA全长序列(FASTA格式),其余12属大多为新确立的属,相关研究较少,且丰度不高,予以排除.这142个属的16S rDNA的序列,构成了本次实验中所用的16S rDNA库.
2.2 引物设计

注: 表中所有引物,属内覆盖度(Cover Rate)均高于80%,且属间特异性值为0,即不与任何环境属序列反向互补.
实验设计了长度在20~25bp,产物长度在200~300bp的引物,要求目标属内覆盖率达到80%以上,与其他属序列均存在差异.排除了两端引物tm差值5℃以上,或具有4bp以上特殊二级结构或连续相同碱基的引物.最终,从Flavobacterium中设计出174对引物,从Pseudomonas中设计出2291 对引物.Shewanella和Rhodobacter缺乏成对的具有特异性的引物,一端使用普通引物进行补全,最终从Shewanella中得到1009对次优引物,从Rhodobacter中得到190对引物.用NCBI的Primer-BLAST进行验证,确定引物特异性后,每个属中选择了一对引物进行后续验证实验(表2).
2.3 样本16S rDNA的提取
实验利用DNeasy Blood & Tissue Kit(QIAGEN,Chatsworth,CA,USA)的操作流程分别提取LeH和NonLeH两个样本中的微生物总DNA,并用16S rDNA的通用引物27F(5’-AGAGTTTGATCCTGGCTCAG-3’)和1492R(5’-TACGGCTACCTTGTTACGACTT-3’)对总DNA进行扩增得到总16S rDNA,50℃加热3min,95℃加热10min.之后95℃变性15s,52℃退火30s,72℃延伸40s,执行30个循环.最后72℃延伸10min.产物用QIAquick PCR Purification Kit(QIAGEN, Chatsworth, CA, USA)回收.
2.4 目标属片段的获取
以回收的PCR产物为模板,分别加入对应的引物.进行PCR扩增,50℃加热3min,95℃加热10min.之后95℃变性15s,60℃退火30s,72℃延伸40s,执行30个循环.最后72℃延伸10min.产物与DL2000 DNA Marker共同进行琼脂糖凝胶电泳后验证长度后,用QIAquick PCR Purification Kit回收.之后,将产物用ABI 3730测序仪测序,验证引物的特异性.
2.5 模板的制作
利用Nanodrop 2000超微量分光光度仪测量目标属片段的吸光度,确定其拷贝数.并梯度稀释成1.0×106~1.0×102/μL,作为标准曲线的模板.同样,测定LeH和NonLeH两个未知样本16S rDNA的吸光度,确定其拷贝数后,梯度稀释至1.0×105/μL,作为未知样本的模板.
2.6 Real-time PCR定量
本实验使用上海捷瑞生物工程有限公司的Power qPCR PreMix(SYBR Green)试剂盒和Roche lightcycler 480实时荧光定量PCR仪进行Real-time PCR.实验分成4个组,分别对应3个致病属和一个辅助检测属,每一组分别为病患部位、非病患部位和5个梯度的标准曲线,并进行3次重复,一共有21个样品.每一个样品中含有PreMix 10μL,前后引物各1μL,DNA模板2μL,水4μL,构成20μL反应体系.混匀之后,按照95℃变性15s,60℃退火20s,72℃延伸18s进行,反应共进行40个循环.之后温度由72℃逐渐上升至95℃,绘制溶解曲线.得到Cp值和溶解曲线,进行相对定量分析.
3 结 果
3.1 琼脂糖凝胶电泳和序列测定
利用总16S rDNA和4个属的特异性引物进行扩增,之后利用琼脂糖凝胶电泳,得到电泳图片(图2).可以看到目标属片段均在250bp左右,与设计目标相符.
对得到的PCR产物片段进行一代双向测序,去除杂峰得到测序结果(表3,具体序列看67页附表1).由于引物扩增了目标属中的多个种,因而结果中有简并碱基出现.利用BLAST工具将片段与NCBI 16S rDNA数据库对应属的序列进行比对.由于BLAST不能识别简并碱基,手工将简并碱基引起的错配消除后,记录错配的位数(表3).Shewanella的F方向测序中,有3个碱基在所有种都有错配,R方向正确匹配,因此怀疑该错配为测序错误.除此之外所有属都有大量种与测序结果完全匹配,说明引物确实正确扩增了对应属的序列.

图2 琼脂糖凝胶电泳图片Fig.2 Picture of agarose gel electrophoresisM: DNA Marker,自上而下分别为2000,1000,750,500,250,100bp; 1. Flavobacterium; 2. Pseudomonas; 3. Shewanella; 4. Rhodobacter
Tab.3 Results of sequencing and BLAST

样本产物长度/bp方向测序长度/bp最少错配位数/个最多错配位数/个Flavobacterium254F11700R18301Pseudomonas212F7602R14401Shewanella235F136310R11502Rhodobacter229F12500R16100
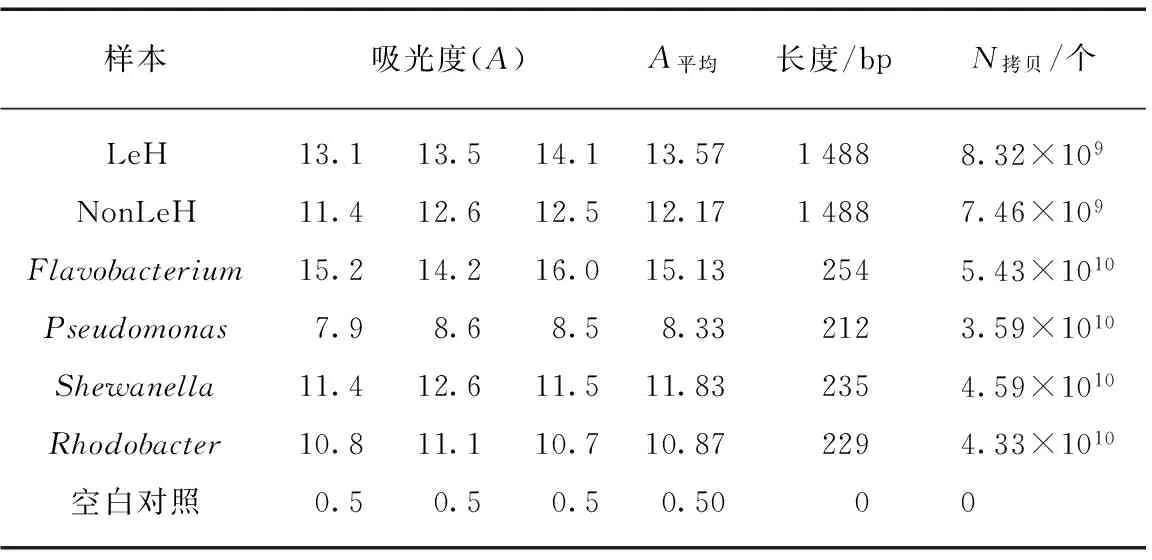
3.2 吸光度测定和拷贝数计算
利用Nannodrop 2000分光光度计测得PCR产物的吸光度,并根据扩增产物的长度估算产物的拷贝数,结果见表4.为保证LeH和NonLeH 2个未知样本的拷贝数为1.0×105/μL,需对其进行5次10倍梯度稀释.
3.3 溶解曲线、扩增曲线和标准曲线
Real-time PCR反应的溶解曲线如图3(看64页)所示.可以看到,4个属的溶解曲线均为单峰,且峰的宽度比较窄.由于引物在属内具有较高的覆盖度,因此PCR扩增得到的产物并不是单一的.然而,由于产物的长度都在200~250bp之间,且16S rDNA的序列本身较保守,因此不同的产物之间也应该仅有几个碱基的细微差别,溶解温度相近,体现在溶解曲线的单峰上.同时,这也说明了Real-time PCR中没有其他的扩增产物生成.Real-time PCR反应的扩增曲线如图4(看64页)所示.
对于每一组,实验对扩增曲线分别设置了基线和阈值,绘制标准曲线(图5,看65页),并确定未知样本的Cp值(每个反应管到达阈值所用的循环数)和拷贝数.
3.4 致病菌的相对定量
对于每个样本的3个重复,实验舍去一个偏差较大的,其他2个求平均值,作为未知样本的结果(表5).最后,与16S rDNA样本的拷贝数做对比,得到4个属占总16S rDNA的百分比,分析结果见表5.根据Real-time PCR反应在LeH和NonLeH两个样本中得到的目标属的Cp值和拷贝数,以辅助检测属Rhodobacter为基准,对3个致病属Flavobacterium、Shewanella和Pseudomonas进行相对定量(表5,看65页).结果表明,在患病样本LeH中,致病属Flavobacterium的相对比例有显著提高,大约为未患病样本NonLeH的两倍.而另外两个致病属Shewanella和Pseudomonas的比例不但没有上升,反而还有一定的下降.由此,我们推断,患病样本的主要致病菌为Flavobacterium.


图3 Real-time PCR溶解曲线Fig.3 Melting curves for Real-time PCR横坐标为反应温度,单位为℃.纵坐标为荧光信号强度的负一阶导数.4个目标属均为单峰,峰较窄,说明产物特异性好.
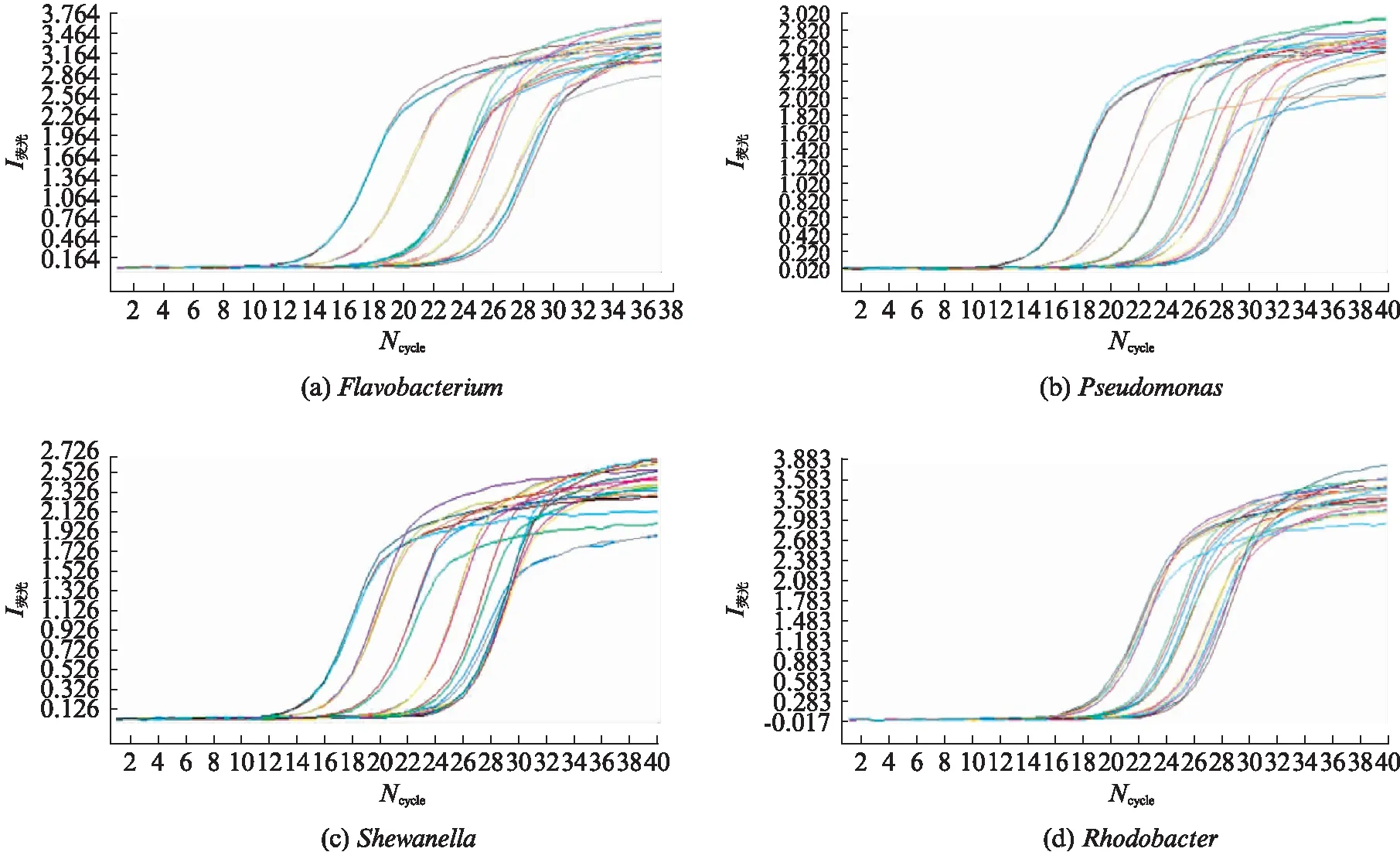
图4 Real-time PCR 扩增曲线Fig.4 Amplification curves for Real-time PCR横坐标为循环数Ncycle,纵坐标为荧光强度I荧光.

图5 Real-time PCR标准曲线Fig.5 Standard curves for Real-time PCR
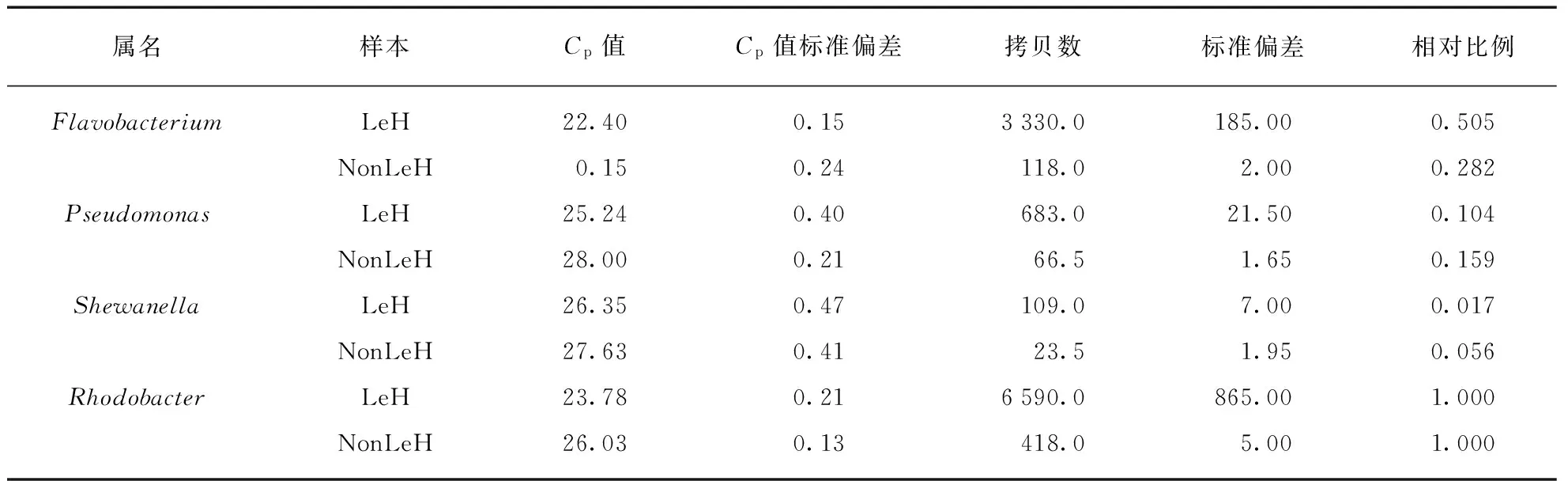
属名样本Cp值Cp值标准偏差拷贝数标准偏差相对比例FlavobacteriumLeH22.400.153330.0185.000.505NonLeH0.150.24118.02.000.282PseudomonasLeH25.240.40683.021.500.104NonLeH28.000.2166.51.650.159ShewanellaLeH26.350.47109.07.000.017NonLeH27.630.4123.51.950.056RhodobacterLeH23.780.216590.0865.001.000NonLeH26.030.13418.05.001.000
4 讨 论
本研究针对属水平特异性引物的设计需要,对16S rDNA在属水平上进行特异性引物设计优化,通过多轮筛选,最终确定需要的引物对,达到在菌群分析、致病菌初期筛查等方面应用的要求.和现有的引物设计思路相比,本研究更加注重于引物的特异性,即从可能的环境属入手,排除可能的错配,达到特异性扩增的目的.
本研究的一个特点是生成了符合特定环境的引物对,即将特异性限制在一定的环境之中.本研究一开始确定了环境中可能存在的其他DNA序列,通过特异性片段的对比,排除这些序列的干扰,这样就确保了PCR反应在这一环境中的特异性扩增.这样的设计思路有以下两点优点:
第一,更容易得到特异性引物.Primer-BLAST中特异性检测在NCBI的数据库中进行,这样的大型数据库数据量大,因此难以找到单一与目标模板匹配的引物对,这也体现在利用引物进行BLAST的时候,引物对往往和许多其他序列匹配,这些序列就会造成错误扩增.本研究只是在具体环境中进行了特异性检测,更容易得到在具体环境中具有特异性的引物.即是引物可以和某些序列结合,但是由于这些序列并未出现在环境中,也不会发生错误扩增,产生干扰.
第二,大量的特异性引物有利于提高引物效率.由于本研究只是对具体环境中可能存在的微生物进行了特异性检测,因此相对于在大型数据库中对所有已知微生物进行的特异性检测,可以得到更多的引物对.这些引物对允许我们对tm差值、二级结构等不利于扩增的因素进行进一步筛选,从而保证了引物效率.同时,本研究考虑到了目标属内的覆盖度,保证了一对引物可以结合目标属中的大部分序列.这一筛选过程是针对菌群分析和致病菌初期筛查等方面的应用设定的,是本研究较为核心的一部分.然而,这一步需要大量的运算,虽然我们有针对性地进行了算法优化,但仍在引物设计过程中占用较多时间,可能需要进一步优化.
我们使用淞江鲈体表菌群进行Real-time PCR分析,验证了引物的特异性和效率.电泳中的条带和测序结果表明,目标条带确实是我们预期的理想产物,具有较好的属内覆盖度和属间特异性.同时,Real-time PCR反应结果也证实该方法设计的引物具有较好的特异性和较高的扩增效率.
我们将本研究使用的各步骤编写为一个小程序(使用MIT软件协议),同时编写了基于Qt的图形界面(http:∥homepage.fudan.edu.cn/zhouyan/primer-maker/),有专门的设置界面对每一步的筛选操作进行参数设置,同时加入了进度显示来估计运行的时长和所需的运行时间.软件输出的结果表示为引物对的形式,也显示了与模板匹配的位置和产物长度.
本软件采用了多线程设计,将“后台数据运算”与“图形化界面”分离,使研究者可以在软件运行时,清晰地看到数据处理的实时进度.输入序列时,本软件采用自动处理的模式,只需要导入全部属的基因信息,并给出一份目标属的清单目录,省去了研究者手动在大量的文件中将目标属和环境属各自归类的麻烦.
查找目标属内保守片段时,本软件采用了自创的“切分”查找算法,极大地提升了查找效率.假设目标属内的某个种,其基因片段长达1000,而需要的特征片段长度为20,若采取传统的“暴力搜索”方法,需要进行检验的字符串有981个.现采用“切分”算法,将这长度为1000的基因串切分为100段长度为10的小串.只搜索这100段小串,对于匹配成功的小串,再向左右延伸,直到达到需要的20长度.这样不会错过任何一个正确的答案,运行速度也有了指数级别的优化提升.输出部分,既支持在软件窗口中进行快速地查看,也支持将结果保存导出为文件.
在实验过程中,我们首先使用16S rDNA的通用引物对样本进行扩增,得到所有样本的16S rDNA序列,这一步和我们目的基因的选择相关.引物设计中,如果采用环境属的全基因组作为原数据,特异性筛选时会去掉大量的结果,部分特异性不够的属无法设计满足要求的引物.于是我们选择所有样本的16S rDNA序列作为原始数据,这样只能保证引物在16S rDNA中的特异性,不能保证在全基因组中的特异性.因此,在实验过程中,要先提取所有样本的16S rDNA作为原始样本.如果想要在环境属全基因组中直接扩增,可以利用BLAST,在NCBI、RDP等数据库中进行检测.如果结果中不含目标属以外的属,且引物在目标属中匹配到了正确的位置,即可以直接在环境属全基因组中使用,不必预先提取目的基因.我们随机选取软件设计出来的一些引物进行了BLAST检测,确实出现某些16S rDNA中特异的引物在环境属基因组中不具有特异性的情况(表7).

表7 一些引物的BLAST检测

附表1 测序结果
致谢及分工: 项目中,徐鹏昊,王焱磊,蔡枫,张亮,窦同海,周雁负责引物设计及优化,何恩明在实验部分提供帮助,罗武松、王金秋负责样本采集.感谢王翌霞的前期工作对本文的启发,感谢杨芳在实验方面给予作者的帮助.
参考文献:
[1] LI X R, MA E B, YAN L Z, et al. Bacterial and fungal diversity in the traditional Chinese liquor fermentation process [J].InternationalJournalofFoodMicrobiology, 2011,146(1): 31.
[2] LOCH T P, FAISAL M. Deciphering the biodiversity of fish pathogenicFlavobacteriumspp. recovered from the great lakes basin [J].DiseasesofAquaticOrganisms, 2014,112(1): 45.
[3] QIN L, ZHU M, XU J. First report ofShewanellasp. andListonellasp. infection in fresh water cultured loach,Misgurnusanguillicaudatus[J].AquacultureResearch, 2014,45(4): 602.
[4] STARLIPER C E, MORRISON P. Bacterial pathogen contagion studies among freshwater bivalves and salmonid fishes [J].JournalofShellfishResearch, 2000,19(1): 251.
[5] RYCHLIK W. OLIGO 7 primer analysis software [J].MethodsMolBiol, 2007,402: 35-60.
[6] YE J, COULOURIS G, ZARETSKAYA I, et al. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction [J].BMCBioinformatics, 2012,13: 134.
[7] COOK VJ, TURENNE CY, WOLFE J, et al. Conventional methods versus 16S ribosomal DNA sequencing for identification of nontuberculous mycobacteria: Cost analysis [J].JournalofClinicalMicrobiology, 2003,41(3): 1010-1015.
[8] COLE J R, WANG Q, FISH J A, et al. Ribosomal Database Project: Data and tools for high throughput rRNA analysis [J].NuclAcidsRes, 2014,42: 633-642.
[9] PAN L D, CAI F, MA Z, et al. Population characteristic and protection of roughskin sculpin in China [J].FishSciTechnolInform, 2010,37: 211-214.
[10] 王翌霞,罗武松,秦志浩,等.利用高通量测序技术探究淞江鲈体表细菌的分类 [J].云南农业大学学报,2016,31(1): 87-94.
[11] IMHOFF J F, TRÜPER H G, PFENNIG N. Rearrangement of the species and genera of the phototrophic “Purple Nonsulfur Bacteria” [J].InternationalJournalofSystematicBacteriology, 1984,34(3) : 340.
猜你喜欢
粉末冶金技术(2021年3期)2021-07-28
传染病信息(2021年6期)2021-02-12
中国生殖健康(2020年2期)2021-01-18
天然产物研究与开发(2019年10期)2019-11-05
科海故事博览·下旬刊(2019年6期)2019-04-16
中国生殖健康(2018年4期)2018-11-06
中国医疗保险(2017年5期)2017-05-17
中国民族医药杂志(2016年2期)2016-05-14
中国康复理论与实践(2015年10期)2015-12-24
现代电生理学杂志(2015年1期)2015-07-18
