
植物基因启动子的克隆及分析的研究进展
2018-05-09 01:19马宝月
中国农业文摘-农业工程 2018年3期
马 倩,马宝月,穆 波,马 慧
(沈阳农业大学生物科学技术学院,沈阳 110161)
启动子在基因转录调控中占有核心地位,决定基因的表达水平,是位于基因的上游、能够活化RNA聚合酶的一段DNA序列。在高等植物中,基因的表达调控过程主要有DNA水平、染色体水平、转录调控、转录后调控、翻译调控和翻译后调控,在这六大调控方式中,转录水平的调控方式是最为主要的,它主要受顺势作用原件与转录因子之间的协调作用。而在启动子区域,有很多能够在转录水平调控过程中具有重要作用顺势作用原件,其通过驱动下游基因的表达来增加植物对非生物胁迫的抵抗能力。所以,对启动子进行克隆,并仔细分析其序列功能对具有重要作用的功能原件和调控区域进行精准的定位。有利于研究基因的调控机制及转录调控模式,并且应用于基因工程从而进行植物遗传改良。
1 克隆启动子的几种常用方法
1.1 常规的PCR技术克隆启动子
利用常规PCR方法,根据已知的启动子序列来设计扩增全长序列的引物,由于该方法操作简便并且快捷,所以,近年来被广泛的应用在克隆序列已知的基因启动子中,但是此方法不能用来克隆未知的启动子序列。
自从1985年PCR技术诞生以来,通过PCR技术来克隆基因启动子便成为了最常见的一种方法。Tao等(2014)根据已发表的玉米ZmRXO1基因的启动子序列设计引物,以玉米的基因组DNA为模板,利用PCR技术扩增反应得到1576 bp的ZmRXO1基因的启动子。苏宁等(2003)以已经发表的水稻中叶绿体的16S rRNA基因的启动子碱基序列为理论依据进行引物的设计,以水稻的叶绿体DNA为模板,利用PCR技术进行扩增反应,发现扩增出的序列与已知的启动子序列同源性高达100%。由此可见,利用常规PCR方法克隆已知序列的启动子,操作简单、便捷,准确度高。但缺点在于不能克隆新的启动子。
1.2 反向PCR
反向PCR(I-PCR,Inverse PCR)是Triglia等(1998)最早提出的在PCR基础上改进的染色体步移的技术。其原理如图1所示:首先使用合适的限制性内切酶将植物的基因组DNA进行酶切反应,然后利用T4连接酶将DNA片段自连,使其形成环状的DNA,然后再以环化的DNA作为反应模板,根据已知序列来设计反向引物,通过PCR扩增反应来获得未知片段。韩志勇等(2001)利用I-PCR技术,以转基因水稻为试验材料,通过克隆技术获得了外源基因的侧翼序列,并且在一周时间内,通过克隆获得的35个水稻转基因株系长度大约在300~750 bp的外源基因的侧翼序列。张晓等(2012)采用I-PCR和TAIL-PCR方法从棉花中克隆到了线粒体atpA双拷贝基因的侧翼序列。该方法高效、快速、稳定、简单便捷,引物设计方便,但是PCR扩增过程容易形成非特异性产物,效率比较低。
1.3 接头PCR
接头PCR技术是一种对植物的基因组DNA已知的序列两侧未知序列进行扩增的技术,是继反向PCR技术之后的又一种克隆启动子的方法,可以用来对已知的基因序列上游启动子的序列进行PCR扩增。其原理为:设计一个长链接头和一个与长链5’端互补的短链接头,需要在短链接头3’端添加一个NH2,以防止聚合酶在促反应时短链进行延伸,再用几种合适的限制性内切酶对基因组DNA进行酶切反应,然后利用T4连接酶把酶切后得到的DNA片段与合成的接头引物相连接,以连接后的产物作为PCR扩增反应的模板,以接头特异性引物(与接头序列互补)和基因特异性引物(与已知基因5’端序列互补)作为上下游引物进行两轮的巢式PCR扩增。

图1 I-PCR原理Triglia等(1998)
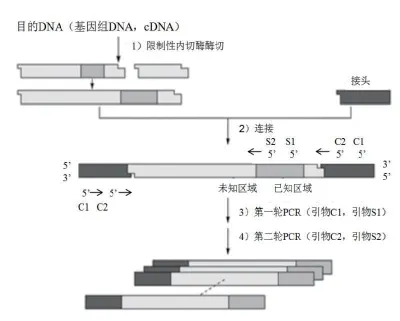
图2 LA-PCR试剂盒流程图Li等(2005)
Xin等(2012)首先利用限制性内切酶对橡树的基因组DNA进行酶切,然后经过两轮的巢式PCR扩增反应后,成功的获得了6个蔗糖转运蛋白基因、1个海藻糖合酶基因和1个海藻糖转化酶基因。根据接头PCR技术的反应原理,宝生物公司推出了LA-PCRTMin vitro Cloning Kit,其用来对基因侧翼的未知序列进行扩增反应,原理如图2所示。Li等(2005)利用该试剂盒在山葡萄中进行扩增,获得了几丁质酶基因VCH3上游的1216 bp启动子序列。该方法操作简单便捷,但是扩增产物特异性较差,扩增产物需要进一步杂交验证。
1.4 热不对称交错PCR
热不对称交错PCR(thermal asymmetric interlaced PCR,TAIL-PCR)是由Liu等(1995)最早提出的,该方法以基因组DNA为模板,由已知目的基因的5’端序列,设计了3个嵌套引物(sp1,sp2,sp3,20 bp左右)和Tm值较低的随机简并引物(AD,14 bp左右)组合,进行3轮PCR反应扩增特异性产物,能够高效快速地扩增未知序列,试验流程如图3所示。Li等(2007)结合了接头PCR技术和TAIL-PCR技术两种扩增方法,以辽宁碱蓬的基因组DNA为模板,克隆获得了SlCMO基因上游的2204 bp的启动子序列。陈军营等(2007)以小麦为试验材料,采用TAIL-PCR方法进行克隆,得到了位于GLP3基因上游的1748 bp处的启动子的碱基序列。利用TAIL-PCR方法进行克隆,克隆前不需要对基因组DNA模板进行酶切,其优点在于能够避免产生环化和连接、扩增的产物特异性强、扩增效率高、灵敏度高,已经在分子生物学克隆未知序列的研究中被广泛应用,但是使用该方法需要较高纯度的模板DNA和引物,扩增条件也比较严格。

图3 Genome Walking Kit的实验流程图Liu等(1995)
1.5 融合引物巢式PCR
融合引物与巢式聚合酶链式反应(fusion primer and nested integrated PCR,FPNI-PCR),是一种在TAIL-PCR方法原理的基础上改进的PCR技术(Wang et al. 2011)。在随机通用引物的5’端连接上接头序列,使第二轮、第三轮的PCR变成了普通PCR,节省了实验时间。根据已知基因5’端序列来设计3个特异性的嵌套引物,并与9个特殊设计的随机简并引物(AD)混合物相配合,进行第一轮的巢式PCR基于随机通用引物去设计两个具有特异性的引物来代替AD,进行第二和第三轮的常规PCR,第二和第三轮的FPNI-PCR是以上一轮的PCR扩增产物稀释25或50倍为模板,再进行PCR扩增,获得扩增产物,试验流程如图4所示。Zhang等(2013)采用FPNI-PCR方法,在日本落叶松中克隆到了LaTCTP基因的上游的启动子序列,利用软件分析结果显示在体细胞胚胎发育过程中存在很多起调控作用的顺式作用元件。李婧等(2016)利用FPNI-PCR技术进行克隆,在大花三色堇中获得了花青素合成酶(VwANS)基因的上游启动子序列,并对实验反应条件进行了优化,实验结果表明模板的稀释倍数是根据简并引物的不同而进行调整,才能扩增到清晰地条带。FPNI-PCR技术克隆已知基因的侧翼序列可以有效的降低非特异性扩增,具有快捷方便、稳定性好、灵敏性高的获得目的序列的优点,是扩增未知片段的有效技术。
2 诱导型启动子的功能研究
启动子本身并不能直接参与基因活动,而是需要通过与转录因子相结合才能发挥其作用。
控制基因表达的时间和表达程度。所以,启动子的功能主要由启动子中与转录因子集合的元件决定。目前,在高等植物诱导型启动子功能的分析与研究中,常用的方法主要包括生物信息学、瞬时表达分析、点突变、稳定表达分析、酵母单杂技术、凝胶阻滞分析(EMSA)、DNase Ⅰ足迹分析等(Behnam et al. 2013),以及近些年应用的RNAi技术等。
2.1 生物信息学分析

图4 FPNI-PCR实验流程图Wang 等(2011)
利用生物信息学软件对克隆获得的启动子序列进行元件的预测与分析,为今后的启动子功能研究提供了依据,同时也促进了启动子的研究效率(Hehl et al. 2001)。目前,能够对启动子的功能元件进行预测和分析的常用的数据库及分析软件有:
2.1.1 Promoter Scan(http://www.bimas.dcrt.nih.gov.molbio/proscan)转录调控的数据库(Prestridge 1995)
其主要功能是进行启动子预测。已经开发了计算机程序PROMOTER SCAN,以识别Pol II启动子序列的百分比,同时仅允许较小的误报率。PROMOTER SCAN目前是最好的三个程序识别灵长类启动子序列,PROMOTER SCAN能够识别大部分新颖性的提案启动子序列,同时保持低的假阳性率。
2.1.2 PLACE(plant cis-acting regulatory DNA elements http://www.dna.affrc.go.jp/PLACE)植物DNA顺式调控元件的数据库(Higo et al.1999)
PLACE能够总结所查数据的当前地位及可利用的工具。PLACE数据库中每个基序的文档包含每个基序的简要定义和描述,以及可用的PubMed ID号和GeneBank登录号的相关文献。用户可以使用网站上的SingalScan程序的顺式作用原件查询序列。结果将以三种形式之一报告。
2.1.3 Promoter 2.0(http://www.cbs.dtu.dk/services/Promoter)顺式作用元件的数据库(Knudsen 1999)
Promoter2.0目前已经被开发为和启动子区域中的序列相互作用的模拟转录因子的进化。它建立在神经网络和遗传算法常用的原则上。
2.1.4 PlantCARE(http://bioinformatics.psb.ugent.be/webtools/plantcare/html/)植物顺式调控元件(Rombauts et al.1999)
PlantCARE是植物顺式调控元件,增强子和阻遏物的数据库。除了在序列上发现的转录基序之外,它还提供了包含完整基因序列的EMBL条目的链接以及基序变成功能的条件的描述。
2.1.5 TRANSFAC(transcriptional regulation from patterns to profiles http://www.gene-regulation.com)转录因子的数据库(Matys et al. 2003)
提供实验确定的转录因子结合位点和位置权重矩阵的最全面的收集。包含转录因子数据,靶基因和调控结合位点的TRANSFAC数据库已被扩展和进一步开发,无论是条目数量以及收集的数据的范围和结构。表达模式的结构域已被引入人类和小鼠的转录因子,使用CYTOMER数据库在解剖结构和发育阶段。
2.1.6 PlantPAN(http://plantpan2.itps.ncku.edu.tw)用于检测转录因子结合位点的数据库(Chow et al. 2016)
植物启动子分析导航器为检测转录因子结合位点(TFBS),相应的TF和其他重要的调控元件(CpG岛和串联重复序列)在植物中的启动子或一组启动子中。目前的PlantPAN版本(2.0版)在76种植物中含有16 960个TF和1 143个TF结合位点的基因。
2.2 点突变
点突变分析可以精确定位核心调控元件的位置,可通过转座子插入突变,或用限制性内切酶酶切特定的功能元件,然后再将插入的突变片段或者启动子的缺失片段转入到植物中确定的特定原件。此方法是在研究启动子的功能元件中经常用到的方法,利用该方法分析获得的元件主要有TATA-box和CAAT-box。(Khuranan et al. 2013)。
2.3 瞬时表达分析
瞬时表达分析主要是将启动子连接到报告基因的上游,然后转化到模式植物中,从而使报告基因能够在短时间内得到表达,而外源基因则不需要整合到植物的基因组中,而是在胁迫条件下,通过对报告基因表达量的检测来分析启动子的活性,(Logemanm et al. 2013;Lu et al. 2013),瞬时表达只能进行定性分析,而不能用作对启动子诱导活性的检测,是对启动子是否具备启动功能进行验证的常用方法。
2.4 稳定表达分析
稳定表达分析的原理是将缺失表达片段与将启动子序列去除的表达载体序列连接构建缺失表达载体,然后通过基因枪法、农杆菌介导法等方法转化模式植物,进而获得能够稳定表达的植株。然后对稳定表达的转基因植株进行胁迫处理,对报告基因的表达量进行检测。根据对报告基因表达量的分析,来判断启动子的诱导活性和组织特异性的调控元件(Esfandiari et al. 2013)。通过构建不同缺失片段表达载体转化植物,可以根据报告基因表达量的高低精确地确定顺式作用元件的具体位置和作用功能(Alzohairy et al. 2013;Creux et al. 2013)。稳定表达分析是对启动子的功能研究中最为常用的一种分析方法,能够确定启动子的不同区段作用元件的功能,能够准确地定量分析报告基因的表达量,实验结果可靠,但是获得稳定表达的转基因材料的周期长。
2.5 酵母单杂技术
酵母单杂交技术(yeast one-hybrid)最初是由Li等在酵母双杂技术的基础上改进的,根据报告基因的表型来分析启动子区域作用元件与转录因子的相互作用的一项技术。由于酵母单杂交技术检测特定的转录因子与顺式作用元件特异性的相互作用的敏感性和可靠性,已被广泛的用于克隆细胞中含量低的、采用生化纯化手段难以纯化的转录因子(文添龙等,2014)。
2.6 凝胶组织分析
凝胶阻滞分析(electrophoretic mobility shift assay,EMSA)是一种能够简单并且快速地检测出蛋白质与特异性DNA序列间结合情况的技术,可以用来对启动子和特定转录因子之间结合情况的检测,从而能够确定出启动子的区域核心作用元件。其作用原理:在凝胶电泳过程中,由于电场正负极的作用,小分子DNA片段向阳极移动的速度比结合了蛋白质的DNA片段的速度更快,因此,根据条带迁移率的快慢程度就能够判断出DNA分子与蛋白质的结合情况。凝胶阻滞分析也是研究启动子的区域功能元件和转录因子之间结合情况的一种重要手段,进而确定出新的作用元件。或者用来验证已知作用元件与转录因子的特异性结合情况。
2.7 DNaseⅠ足迹分析
DNase Ⅰ足迹分析法(DNase footprinting)也是用于研究顺式作用元件与蛋白质相互作用的一种方法,该方法能够精确地确定与蛋白质结合的DNA片段长度及序列(Galas et al. 1978)。其基本原理是用DNase I部分消化已进行单链末端标记的待测双链DNA,在变性聚丙烯酰胺凝胶上形成以相差一个核苷酸为梯度的DNA条带;当DNA片段与其蛋白质特异性结合后,结合蛋白的区域将会受到蛋白的保护,避免受到DNase I的攻击,因此形成切割梯中的空白区域,再经过DNA化学测序法,就可预测到该结合区的碱基序列。
3 结语与展望
在植物基因的表达过程中,调控基因表达的途径有很多,其中转录水平的调控起着非常重要的作用。在转录水平上,由于启动子区域的顺式作用元件和转录因子之间相结合,共同参与调控功能基因的表达。因此对于诱导型启动子来说,在克隆和功能分析的研究越来越受到广泛的关注。目前对诱导型启动的研究技术初期阶段,将胁迫诱导型启动子通过基因工程的手段转入植物,增强植物的抗逆性,但其分子机制却不清楚。在本文中,简要的介绍了克隆启动子的几种常用的方法以及功能分析的方法,随着分子生物学技术的逐步发展,将会出现更多的克隆启动子的方法和应用于启动子顺式作用元件分析的技术,将会发现越来越多的功能元件和转录因子,从而提高植物的基因调控过程水平的能力。对了解基因的表达调控模式奠定基础,为提高转基因植物的表达效率提供重要的依据。
[1] Tao Y,Wang F T,Jia D M, et al. Cloning and Functional Analysis of the Promoter of a Stress-inducible Gene(ZmRXO1)in Maize[J]. Plant Mol Biol,2014,33(2):1-9.
[2] Triglia T,Peterson M G,Kemp D J,et al.A procedure for in vitro amplification of DNA segments that lie outside the boundaries of known sequences,Nucleic Acids Research[J].1998,16(16):81-86.
[3] Han Z Y,Wang X Q,and Shen G Z,Cloning of foreign gene’s flanking sequences in transgenic rice by inverse PCR[N].Shanghai Nongye Xuebao(Acta Agriculture Shanghai),2001,17(2):27-32.(韩志勇,王新其,沈革志,PCR克隆转基因水稻的外源基因旁侧序列[N]. 上海农业学报,2001,17(2):27-32).
[4] Zhang X,zhang R,Sun G Q,et al. High efficiency genome walking method for flanking sequences of cotton mitochondrial double-copy atpA gene based on optimized inverse PCR and TAIL-PCR[N]. Shengwu Gongcheng Xuebao(Chinese Journal of Biotechnology),2012,28(1):104-115.(张晓,张锐,孙国清,史计,孟志刚,2012,优化的反向PCR结合TAIL-PCR法克隆棉花线粒体atpA双拷贝基因及其侧翼序列[N].生物工程学报,2012,28(1):104-115).
[5] Xin L S,YANG,et al. An Improved Promotercloning Method Based on Adaptor-PCR and Its Application in Rubber Tree[J].Bulletin of Botanical Research,2012,32(3):296-303.
[6] Li H Y,Qi J,Shu H R,et al. Isolation and characterization of a chitinase gene VCH3 promoter from grapevine(Vitis amurensis)[J]. Plant Physiol Mol Biol,2005,31(5):485-491.
[7] Liu Y G and Whittier R F,et al. Thermal asymmetric interlaced PCR:automatable amplification and sequencing of insert end fragments from P1 and YAC clones for chromosome walking[J]. Genomics,1995,25(3):674-681.
[8] Li Q L,Yin H,Li D,et al. Isolation and Characterization of CMO Gene Promoter fromHalophyte Suaeda liaotungensis K[J].Journal of Genetics and Genomics,2007,34(4):355-361.
[9] Chen J Y,Sun P,Wang D Q,et al. An Improved TAIL-PCR Method for Cloning of GLP3 Gene Promoter from Wheat Genomic DNA[J],Zhiwu Shenglixue Tongxun(Plant Physiology Communications),2007,43(4):754-758.(陈军营,孙佩,王德勤,陈新建,2007,一种改良的克隆GLP3基因启动子的TAIL-PCR技术,植物生理学通讯,43(4):754-758).
[10] Wang Z,Ye S,Li J,et al.Fusion primer and nested integrated PCR(FPNI-PCR):a new high-efficiency strategy for rapid chromosome walking or flanking sequence cloning[J].BMC Biotechnol,2011,11:109.
[11] Zhang L F, Li W F, Han S Y,et al. cDNA cloning,genomic organization and expression analysis during somatic embryogenesis of the translationally controlled tumor protein(TCTP)gene from Japanese larch(Larix leptolepis)[J].Gene,2013,529(1):150-158.
[12] Li J,Zeng Y,Gong S,Li T G, et al.Optimization of Pansy FPNI - PCR Reaction System[J].Jiangsu Nongye Kexue(Jiangsu Agricultural Science),2016,45(5):72-75.(李婧,曾媛,龚胜,李霆格,杨文汉,王健,2016,大花三色堇FPNI-PCR反应体系的优化[J].江苏农业科学,2016,45(5):72-75).
[13] Behnam B,Luchi S,Fujita M,et al.Characterization of the promoter region of an Arabidopsis gene for 9-cisepoxycarotenoid dioxygenase involved in dehydration-inducible transcription[J].DNA Res,2013,20(4):315-324.
[14] Hehl R and Wingender E,Database-assisted promoter analysis[J].Trends in Plant Science,2001,6(6):251-255.
[15] Prestridge D S,et al. Predicting Pol II promoter sequences using transcription factor binding sites[J].Journal of Molecular Biology,1995,249(5):923-932.
[16] Higo K,Ugawa Y,Iwamoto M,et al.Plant cis-acting regulatory DNA elements(PLACE)database[J]. Nucleic Acids Res,1999,27:297-300.
[17] Knudsen S,Promoter2.0:for the recognition of PolII promoter sequences[J].Bioinformatics,1999,15(5):356-361.
[18] Rombauts S,Déhais P,Van M M,Rouzé P,et al. PlantCARE,a plant cis-acting regulatory element database[J].Nucleic Acids Research,1999,27(1):295-296.
[19] Matys V, Fricke E, Geffers R,et al.TRANSFAC:transcriptional regulation,from patterns to profiles[J].Nucleic Acids Res,2003,31(1):374-378.
[20] Chow C N,Zheng H Q,Wu N Y,et al.Plant PAN 2.0:an update of plant promoter analysis navigator for reconstructing transcriptional regulatory networks in plants[J].Nucleic Acids Research,2016,44(1):1154-1160.
[21] Khurana N,Chauhan H,Khurana P,et al.Wheat chloroplast targeted sHSP26 promoter confers heat and abiotic stress inducible expression in transgenic Arabidopsis Plants[J]. Plos One,2013,8(1):e54418.
[22] Logemann E,Brikenbihl R P,Rawat V,et al.Functional dissection of the PROPEP2 and PROPEP3 promoters reveals the importance of WRKY factors in mediating microbe-associated molecular pattern-induced expression[J].New Phytologist,2013,198(4):1165-1177.
[23] Lu Y,Chen Y,Wu Y,et al. Directly Transforming PCR-Amplified DNA Fragments into Plant Cells Is a Versatile System That Facilitates the Transient Expression Assay[J].Plos One,2013,8(2):e57171.
[24] Esfandiari E,Jin Z,Abdeen A,et al.Identification and analysis of an outerseed-coat-specific promoter from Arabidopsis thaliana[J]. Plant Molecular Biology,2013,81(1):93-104.
[25] Alzohairy A M,Mac Donald M H,Matthews B F,et al. The pJan25 vector series:an enhancement of the Gateway-compatible vector pGWB533 for broader promoter testing applications[J].Plasmid,2013,69(3):249-256.
[26] Creux N M,Bossinger G,Myburg A A, et al. Induced somatic sector analysis of cellulose synthase(CesA)promoter regions in woody stem tissues[J].Planta,2013,237(3):799-812.
[27] Wen T L,Liu X M,Ji Y P,et al. Research Progress of Stress-induced Promoter in Higher Plant[N]. Xibei Zhiwu Xuebao(Acta Bot. Boreal),2014,34(1):206-204.(文添龙,刘雪梅,冀亚萍,俞嘉宁, 高等植物胁迫诱导型启动子的研究进展[N].西北植物学报,2014,34(1):206-204).
[28] Galas D J,Schmitz A,et al.DNase footprinting a simple method for the detection of protein-DNA binding specificity[J]. Nucleic Acids Research,1978,5(9):3157-3170.
猜你喜欢
广东药科大学学报(2022年3期)2023-01-04
生物学通报(2022年1期)2022-11-22
环球时报(2022-09-20)2022-09-20
中国特种设备安全(2022年1期)2022-04-26
中国种业(2021年11期)2021-11-25
今日农业(2020年24期)2020-12-15
生物学教学(2019年3期)2019-03-22
中国核电(2017年2期)2017-08-11
小资CHIC!ELEGANCE(2015年14期)2015-09-23
小资CHIC!ELEGANCE(2015年15期)2015-09-01
