
磁分散固相萃取-高效液相色谱法测定牛奶中5种禁用喹诺酮类药物
2018-05-05 01:30:45刘艳丽杜晓慧冯艺洋赵雅妮常银霞
分析科学学报 2018年2期
刘艳丽, 吴 昊, 杜晓慧, 冯艺洋, 赵雅妮, 常银霞
(山西师范大学化学与材料科学学院,山西临汾 041004)
喹诺酮类药物(Quinolones,QNs)是一类重要的广谱抗菌化合物,被广泛用于人类和动物的传染病治疗[1 - 3],其中一部分可被代谢和排泄掉,剩余的则残留在人类和动物体中[4]。这些药物残留将会威胁到人类和动物的健康[5 - 7]。中华人民共和国农业部第2292号公告声明,不允许洛美沙星、氧氟沙星、培氟沙星、诺氟沙星用于食物源性动物的疾病治疗。美国食品药品监督管理局也于2005年7月29日宣布禁止使用恩诺沙星进行家禽细菌感染的治疗。但目前还没有一种可以同时分析牛奶中上述5种禁用喹诺酮类药物的方法。所以,建立一种可以同时对牛奶中这5种禁用痕量喹诺酮类药物进行检测的方法至关重要。
通常牛奶中QNs含量在痕量水平,因此需要对药物进行预富集来降低牛奶中其他成分对药物检测的影响。常见的预富集方法有分散液-液微萃取[8]、固相萃取[9]、磁分散固相萃取[10]。对比这3种方法,由于磁分散固相萃取过程可以通过外加磁场分离磁性吸附剂与样品溶液,所以磁固相萃取具有萃取过程简单、耗时短、环境友好等优势。本文运用一步溶剂热法合成了磁性还原氧化石墨烯材料(Fe3O4@RGO),并用于萃取牛奶样品中的喹诺酮类药物,对影响萃取效率的因素,如样品溶液的pH、吸附剂、解吸剂、吸附和解吸时间等进行了优化。并结合高效液相色谱-荧光检测器对牛奶中5种禁用喹诺酮类药物进行了分析测定,获得较好结果。
1 实验部分
1.1 主要仪器与试剂
Waters 1525液相色谱仪(美国,Waters公司),配1525双泵系统和2475多波长荧光检测器;KH 2200DV超声波清洗器(昆山禾创超声波仪器有限公司);pHS-3C酸度计(上海精科)。
洛美沙星(LOM)、诺氟沙星(NOR)、氧氟沙星(OFL)、培氟沙星(PEF)、恩诺沙星(ENR)标准品纯度均在99.0%以上,由中国兽医药品监察所购得。其100 mg/L标准溶液由10 mg标准品溶于100 mL甲醇(色谱纯)而得,冷藏于4 ℃备用。1 mg/L混合标准溶液由各标准溶液和色谱纯的甲醇配制而成。
甲醇、乙腈为色谱纯(北京百灵威科技有限公司);H3PO4为色谱纯(日本,Tokyo Chemical Industry公司);FeCl3为优级纯(美国,Sigma公司);乙二醇、乙醇、三乙胺、丙酮均为分析纯(天津科密欧化学试剂有限公司);乙二胺、NaAc为分析纯(北京百灵威科技有限公司)。实验用水由Milli-Q纯水仪制备。
1.2 样品预处理
样品为从本地超市购买的牛奶。将1 mL牛奶样品分散在5 mL乙腈中,以4 000 r/min离心15 min,然后把上清液转移在干净的棕色玻璃瓶中,并将其冷藏于4 ℃备用。
1.3 色谱条件
色谱柱:Gemini C18柱(250×4.6 mm,5 μm;广州菲罗门);柱温箱:30 ℃;流动相:A相为乙腈,B相为0.25 mmol/L磷酸溶液(先用0.2 mol/L NaOH溶液调到pH=3.0,再用三乙胺调到pH=5.7);洗脱条件:0~10 min,13%~20%B;10~20 min,20%~50%B;20~21 min,50%~13%B;21~30 min,13%B;流速:1.0 mL/min;进样量:10 μL;检测波长:激发和发射波长分别为272 nm和425 nm。
1.4 磁性材料的制备
制备了6种氧化石墨烯(GO)含量不同的磁性材料,制备方法如下:分别准确称取0、75.00、115.74、156.48、197.22、237.96 mg GO于盛有25 mL乙二醇、750 mg FeCl3的烧杯中,磁力搅拌,然后加入1.2 g NaAc,继续搅拌30 min,最后转移到50 mL的聚四氟乙烯内胆中.于200 ℃反应12 h。所得磁性材料用无水乙醇和水洗涤数次,然后50 ℃下真空干燥10 h,分别标记为S1、S2、S3、S4、S5、S6。
1.5 萃取过程
首先往50 mL样品溶液中加入适量磁性材料,超声10 min使分析物与磁性材料充分接触。之后用一块磁铁将磁性材料固定在比色管底部并倒掉上清液。然后加0.3 mL无水乙醇和0.1 mL pH=12.0的B-R缓冲溶液,超声6 min分散磁性材料,再用小磁铁收集磁性材料,用0.45 μm的微孔滤膜过滤解吸液,取10 μL注入高效液相色谱仪进行分析。
2 结果与讨论
2.1 磁性材料的表征
用X-射线衍射、扫描电镜和振动样品磁强计对磁性材料进行了表征。X-射线衍射结果表明(图略)所制备磁性材料,在(533)处有还原氧化石墨烯(RGO)的特征峰,在(220)、(311)、(400)、(422)、(440)、(511)有Fe3O4的特征峰;从扫描电镜图(图1)可以看出Fe3O4纳米颗粒的粒径均匀,直径约为30 nm,很好的负载在RGO上,随着GO用量的增加,Fe3O4在RGO上的分布越来越稀疏;磁滞曲线图见图2,结果表明磁性材料磁饱和值大于40 emu/g,说明所制备的磁性材料在外磁场的作用下很容易被分离。

图1 S1、S2、S3、S4、S5和S6的扫描电镜(SEM)图Fig.1 SEM images of S1,S2,S3,S4,S5 and S6
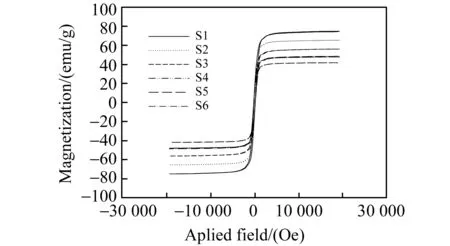
图2 S1、S2、S3、S4、S5和S6的磁滞曲线Fig.2 Magnetization curves of S1,S2,S3,S4,S5 and S6
2.2 磁性材料用量的选择
合适的吸附剂对吸附条件的优化是至关重要的。我们研究了6种GO含量不同的磁性材料S1、S2、S3、S4、S5、S6,结果见图2。由图2可以看出,S3对诺氟沙星的萃取效果没有S2好,但对其余4种禁用喹诺酮类药物的萃取效果比另外的5种磁性材料均好。这可能是由于磁性材料对这几种喹诺酮类药物的萃取是Fe3O4与RGO的协同作用所致,当它们的量呈一定的比例时,磁性材料对药物的萃取效果最佳,当Fe3O4或RGO任意一种含量增大都不利于对药物的萃取。所以在接下来的实验中均以磁性材料S3为吸附剂。
另外,在2~10 mg范围内我们对磁性材料量的影响进行了研究。结果(图3)显示萃取效率随着磁性材料用量的增加不断增强,当磁性材料用量大于7 mg时萃取率开始缓慢下降,这可能是由于磁性材料过多未能完全分散在样品溶液和解吸液中。因此,吸附剂的用量定为7 mg。
2.3 溶液pH的影响
溶液pH影响QNs的存在形式和磁性材料的表面电荷,对萃取效果产生显著的影响。在pH=2.0~12.0范围内研究了pH对萃取效率的影响,如图4。结果表明在pH=2.0时可以获得最大的萃取效率。水溶液中QNs的pKa值为5.66~8.56,表明水溶液中的QNs以阳离子、阴离子、中性分子形式存在[11 - 13]。所以,在pH=2.0时,QNs以阳离子形式存在。因此,较高的萃取效率可能是由于磁性材料表面含氧基团的负电荷与QNs所带的正电荷之间的静电作用。另外还研究了不同缓冲体系(乙酸盐、柠檬酸盐、邻苯二甲酸氢钾、磷酸盐和三酸缓冲溶液)对萃取率的影响,结果表明三酸缓冲溶液(pH=2.0)萃取效果最佳。

图3 磁性材料的选择Fig.3 Selection of magnetic material

图4 溶液pH的影响Fig.4 Effect of solution pH
2.4 解吸剂种类、体积和pH的影响
为了得到一个好的萃取率,选择一个适当的解吸液是相当重要的。我们考察了甲醇、乙醇、丙酮、乙腈对萃取效果的影响,结果(图5)表明4种溶剂均可将分析物从磁性材料上解吸下来,但乙醇的解吸效果最好。另外,我们在pH=2.0~12.0范围内对解吸液的pH进行了研究。结果表明pH=2.0~6.0范围内,随着解吸液pH值的增大萃取率缓慢减小;pH=6.0~12.0范围内,随着解吸液pH值的增大萃取率迅速增大,pH为12.0时,萃取率达到最大。所以解吸液的最佳pH值为12.0。同时,我们对解吸液中有机溶剂和缓冲溶液的体积比进行了优化,结果表明最佳体积比为3∶1。
2.5 吸附与解吸时间的影响
为了评估吸附时间对萃取率的影响,在2~9 min的范围内对吸附时间进行了研究。结果(图6)表明,7 min时5种禁用喹诺酮类药物都达到吸附平衡,因此吸附时间设定为7 min。同时,在1~8 min范围内,研究了解吸时间对萃取率的影响,结果表明4 min为最佳的解吸时间。
2.6 磁性材料的回收利用
我们对磁性材料进行了回收利用。分析物从磁性材料上解吸下来后将磁性材料洗干净并干燥,然后再进行吸附-解吸循环,结果发现第11次吸附-解吸后的萃取率与第1次的相比变化率小于5%,所以所制

图5 解吸剂的选择Fig.5 Selection of desorption solvent

图6 吸附时间的影响Fig.6 Effect of extraction time
备的磁性材料具有良好的耐受度,可以重复使用10次,这极大的降低了样品前处理成本。
2.7 方法分析性能
在最佳条件下对加标空白样品进行分析,所建立方法性能参数列于表1。方法检测限在1~4 ng/L之间,日间和日内相对标准偏差(RSD)都低于5%,说明此方法灵敏度高、重现性好。同时也给出了富集倍数等参数,见表1。

表1 方法的性能参数
a:Extraction recovery;b:Enrichment factor.
2.8 样品分析
用所建立的方法对3种牛奶样品进行了分析,结果列于表2。这3种牛奶样品中均无这5种禁用喹诺酮类药物残留。加标回收率在86.5%~104.1%之间,相对标准偏差小于5%,表明所建立的方法是检测牛奶样品中禁用喹诺酮类药物残留的有效方法。
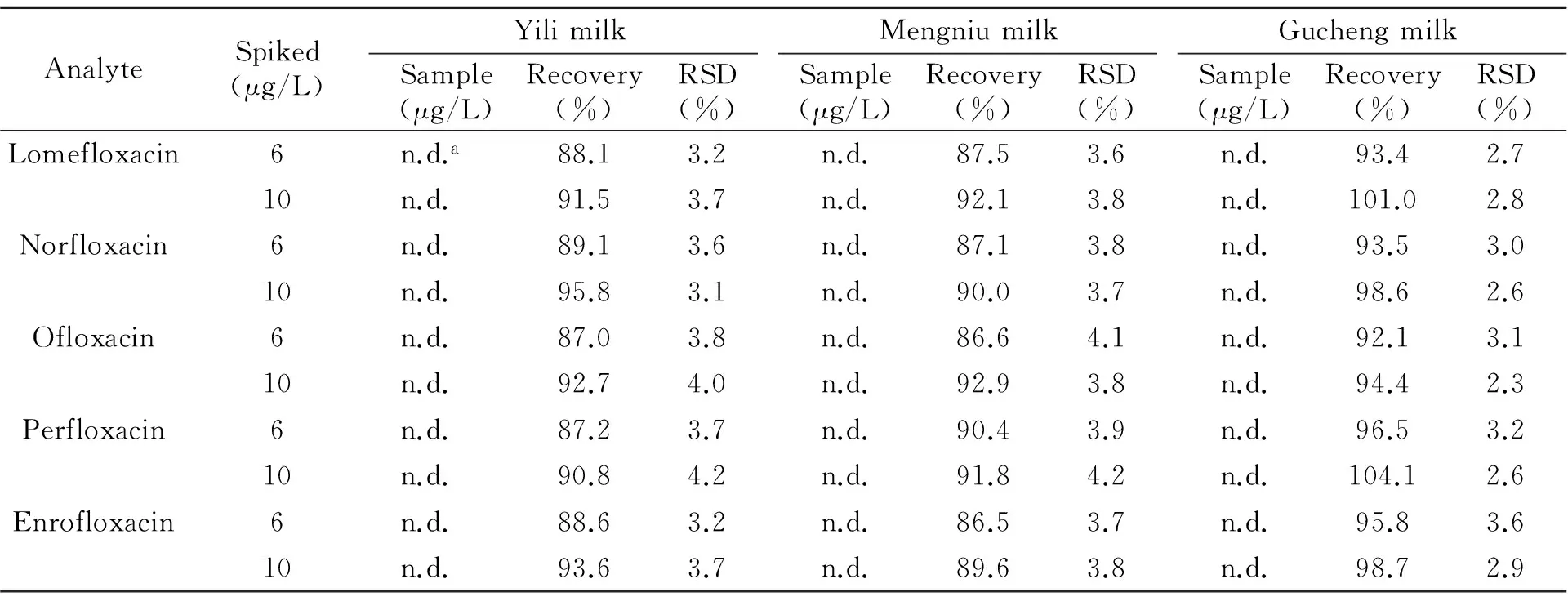
表2 加标牛奶的测定
a:Not detected.
3 结论
提出磁固相萃取-高效液相色谱·荧光检测方法,用于牛奶中禁用喹诺酮类药物测定。Fe3O4@RGO作为吸附剂,避免了离心过程,大大地缩短了实验时间。此方法具有较高的灵敏度。加标回收率为86.5%~104.1%,相对标准偏差小于5%,说明此方法可用于牛奶中5种禁用喹诺酮类药物洛美沙星、诺氟沙星、氧氟沙星、培氟沙星、恩诺沙星的检测。
参考文献:
[1] Samuelsen O B.Aquaculture,2006,255:55.
[2] Wang Y,Yu K,Wang S H.Spectrochimica Acta Part A:Molecular and Biomolecular Spectroscopy,2006,65:159.
[3] He X T,Deng M C,Wang Q,et al.Aquaculture,2016,458:38.
[4] Junker T,Alexy R,Knacker T,et al.Environmental Science & Technology,2006,40:318.
[5] Wang G N,Yang K,Liu H Z,et al.Analytical Methods,2016,8:5511.
[6] Picó Y,Andreu V.Analytical and Bioanalytical Chemistry,2007,387:1287.
[8] LI X S,XU L D,HU X Z,et al.Journal of Analytical Science(李小水,许丽丹,胡西洲,等.分析科学学报),2013,29(30):297.
[9] LIU Y T,LI L,XU C J,et al.Journal of Analytical Science(刘永涛,李乐,徐春娟,等.分析科学学报),2017,33(1):6.
[10] Chen L G,Zhang X P,Xu Y,et al.Analytica Chimica Acta,2010,662:31.
[11] Jiménez-Lozano E,Marqués I,Barrón D,et al.Analytica Chimica Acta,2002,464:37.
[12] Ferdig M,Kaleta A,Buchberger W,et al.Journal of Chromatography A,2004,1047:305.
[13] Su S C,Chang M H,Chang C L,et al.Journal of Food and Drug Analysis,2003,11(2):114.
猜你喜欢
化工管理(2022年13期)2022-12-02 09:21:52
世界最新医学信息文摘(2021年12期)2021-06-09 08:36:56
能源工程(2021年1期)2021-04-13 02:05:50
汽车电器(2019年9期)2019-10-31 07:51:12
国外医药(抗生素分册)(2016年5期)2016-07-12 14:25:37
国外医药(抗生素分册)(2016年2期)2016-07-12 14:25:01
中国卫生标准管理(2015年25期)2016-01-14 09:29:24
科学中国人(2015年25期)2015-02-28 09:15:34
技术与教育(2014年1期)2014-04-18 12:39:16
食品工业科技(2014年15期)2014-03-11 18:17:22
