
应用16S rRNA测序法对过敏性鼻炎患者鼻前庭菌群组成分析
2018-04-24 05:48:27王笛陈昊徐梦雅李子豪单修祺郑兰艳
微生物学杂志 2018年4期
王笛,陈昊,徐梦雅,李子豪,单修祺,郑兰艳
中国医科大学 基础医学院病原生物学教研室,辽宁 沈阳 110122
过敏性鼻炎(AR)是一种耳鼻喉科常见的呼吸道变态反应性疾病,虽不危及生命但却严重影响人们的生活质量。截止到2001年,过敏性鼻炎已累及全球近四分之一的人口[1]。因此,对其致病原因的探讨尤为重要。过敏性鼻炎的致病因素有很多,如花粉、动物皮毛和霉菌[2],且与遗传因素关系密切[3]。近年来,鼻腔中的微生物组成与过敏性鼻炎的关系也受到了人们的密切关注[4]。有学者提出慢性鼻窦炎中的大多数常见菌可能与AR发病具有正相关性[5-6]。但这些研究都依赖于传统的培养方式,因此无法反应鼻腔微生物的真实组成。近年来二代高通量测序技术的发展,为我们分析鼻腔微生物的组成与过敏性鼻炎的相互关系提供了很好的研究平台。本研究采用16S rRNA高通量测序法分析过敏性鼻炎患者鼻前庭微生物组成与正常人的差异,以期揭示过敏性鼻炎患者鼻前庭微生物的组成变化与过敏性鼻炎的发病以及疾病进展的关系,为过敏性鼻炎的个性化诊疗提供新的思路。
1 材料与方法
1.1材料
1.1.1主要试剂 鼻前庭拭子培养管(江苏天力),总DNA提取试剂盒QIAamp DNA Mini Kit(QIAGEN,Hilden,Germany)。
1.1.2研究对象 受试者分两组,试验组、对照组各15人,无性别或年龄偏倚。所有样本均取自沈阳市第八人民医院,历时3个月。考虑到各种可能造成鼻前庭菌群变化的因素,受试者入组标准包括:(1)年龄在20~50岁;(2)3个月内未服用抗生素;(3)不吸烟;(4)未接受任何口腔治疗或外科手术;(5)无怀孕或哺乳期妇女;(6)无系统性疾病。试验组均具有典型症状,临床诊断为过敏性鼻炎,并处于临床发作期。
1.2方法
1.2.1标本采集 取出培养管中的无菌棉签,快速轻柔地旋转擦拭鼻前庭部,每侧各2次,并立即置于-20℃保存。
1.2.2克隆与测序 根据QIAamp DNA Mini Kit使用说明提取微生物总DNA。PCR扩增16S V3-V4可变区,将扩增产物用HiSeq 300PE测序。
1.2.3生物信息学分析 利用Qiime生物信息平台分析下机数据,高质量数据以97%序列相似性[7]进行OTU(operation taxonomy units)聚类。随后进行物种注释及丰度分析。α多样性分析(alpha diversity)揭示样品中的物种构成,β多样性分析(beta diversity)可挖掘样品之间的差异[8]。
1.2.4统计学分析 本实验使用两样本双侧t检验来比较过敏性鼻炎患者与健康人群鼻前庭部菌群的差别,按照α=0.05的水准来进行假设检验统计意义的判定。
2 结 果
2.1alpha多样性 稀疏曲线分析OTU在过敏性鼻炎患者样本内达到132时接近饱和,在健康人体样本内达到141接近饱和,且两组曲线接近,意味着两组鼻腔群落的丰度接近,两组样本测序深度足够(图1A)。Chao1和ACE丰度指标进一步验证了如上观点(图1B)。在本研究中菌群的均匀度我们用Simpsoneven指数进行了分析(图1C)
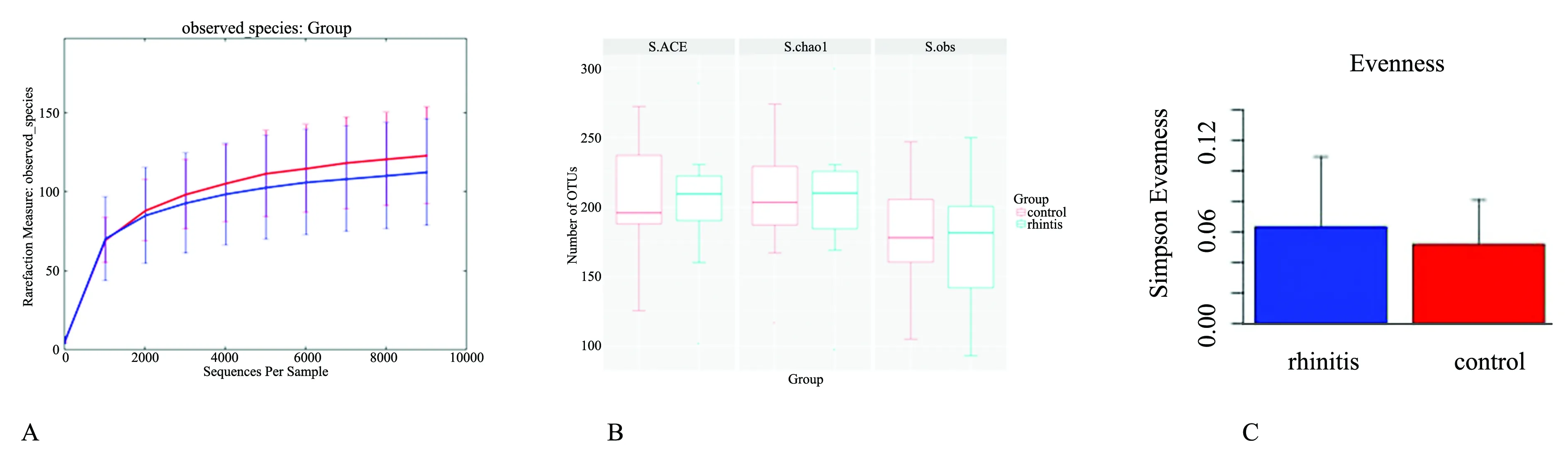
注:A:稀释曲线;横轴显示通过测序获得的序列的数量,纵轴显示在97%序列间相似性的水平下的单元数,为采用的测序工作表示物种数量的近似值,水平线代表不同的OTU±标准误差的平均数(SE)。B:箱线图显示了AR组和对照组使用三种不同的丰度指数OTUs的平均总数:Chao1(S.Chao1)、ACE(S.ACE)和观察到的物种的度量(s.obs)。C:菌种的均匀度经Simpsoneven指数分析,鼻炎组和健康对照组差异无统计学意义(t=0.715,P=0.481)。
图1两组研究对象alpha多样性分析
2.2组间beta多样性 通过加权UniFrac距离测量的方式进行主成分分析(PCoA)和盒子图对过敏性鼻炎患者和健康人群菌群进行了多样性分析。实验基于weighted unifrac distance的多变量方差分析来比较过敏性鼻炎患者和健康人群两组的主成分结构。三维模式显示两组主成分差别较大(图2A)。多变量方差分析的盒子图结果证实了过敏性鼻炎患者鼻前庭菌群与健康对照组差异较大(t=-2.84,P=0.005),且健康组的样本数据相似度很高(图2B),以上结果表明过敏性鼻炎患者组鼻腔菌群样本之间差异较大,即患者的鼻腔菌群异质性高。
2.3菌群组成结构的比较 在菌群门的水平上,过敏性鼻炎患者鼻腔中菌群按丰度递减依次为:变形菌门(Proteobacteria)(55.37%),厚壁菌门(Firmicutes)(25.22%),放线菌门(Actinobacteria)(8.29%),蓝藻(Cyanobacteria)(6.80%),拟杆菌门(Bacte-roidetes)(3.00%),梭杆菌门(Fusobacteria)(0.86%),TM7(0.17%),嗜热菌门(Thermi)(0.12%),酸杆菌门(Acidobacteria)(0.11%),绿弯菌门(Chlo-roflexi)(0.04%)。而健康受试者中,鼻腔中菌群按丰度递减依次为变形菌门(38.46%),厚壁菌门(32.44%),放线菌门(26.40%),拟杆菌门(1.72%),蓝藻(0.63%),酸杆菌门(0.13%),嗜热菌门(0.09%),梭菌门(0.08%),绿弯菌门(0.04%),TM7(0.02%)。过敏性鼻炎患者较健康受试者,厚壁菌门、放线菌门丰度有所减少,而变形菌门、梭杆菌门、拟杆菌门、蓝藻、TM7、嗜热菌门、酸杆菌门丰度较健康受试者相对增加,其中放线菌门(t=-3.184,P=0.004)和梭杆菌门(t=2.107,P=0.046)在试验组与对照组间差异有统计学意义(图3A)。在菌群属的研究水平上,试验组与对照组之间有明显差异的属有13种,包括黄单胞菌科(Xanthomonadaceae)中的一个未注释的菌属(t=-4.0740,P=0.0004)、棒状杆菌属(Corynebacterium)(t=-3.1640,P=0.004)、嗜胨菌属(Peptonophilus)(t=-3.0289,P=0.0057)、厌氧球菌属(Anaerococcus)(t=-2.9675,P=0.0067)、大芬戈尔德菌属(Finegoldia)(t=-2.8727,P=0.0113)、丙酸杆菌属(Propionibacterium)(t=-2.7430,P=0.0113)、短杆菌属(Brachybacterium)(t=-2.3448,P=0.0276)、希瓦菌属(Shewanella)(t=2.3308,P=0.0285)、微小单胞菌属(Parvimonas)(t=2.1943,P=0.0381)、考克菌属(Kocuria)(t=-2.1766,P=0.03656)、鞘脂单胞菌科(Sphingomonadaceae)科中的一个未注释的菌属(t=2.1030,P=0.0461)、嗜盐单胞菌属(Halomonas)(t=2.0763,P=0.0487)和叶瘤菌属(Phyllobacterium)(t=2.0718,P=0.0491)(图3B)。图中只显示了菌属丰度不低于0.1%的9个差异明显的菌属,其他低丰度的差异明显的菌属未在图中显示。
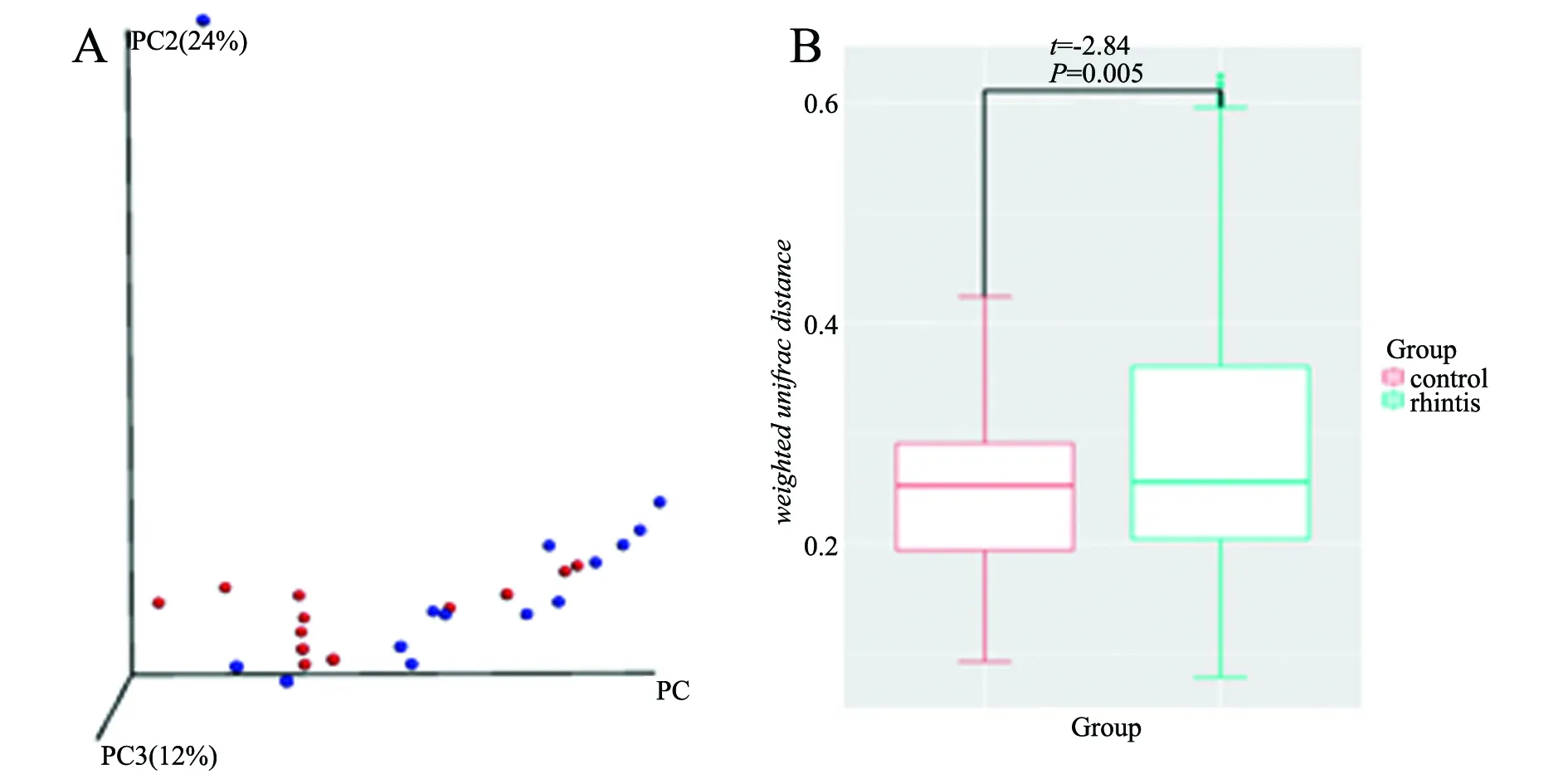
注:A:通过加权UniFrac距离测量的方式进行主坐标分析(PCoA),主坐标分析由三维模式表现,同时在97%序列一致性条件下进行序列聚类。根据细菌组成的26个样品的空间分布,包括AR组(蓝圈)和对照组(红圈);每一点对应一个微生物群落,颜色指示它的类别。B:箱线图显示组内和组间差异性比较得出的平均加权UniFrac距离;通过UniFrac距离和参数t检验证明它们存在显著的差异。
图2两组研究对象beta多样性分析
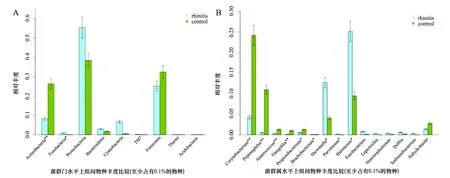
注:A:在门层次上比较AR组和健康对照组的菌群组成差异;Y轴代表菌群的相对丰度,X轴代表各种菌群;单星号表示差异显著,P<0.05,双星号代表差异非常显著,P<0.01。B:在菌属层次上比较AR组和对照组的菌群组成差异;Y轴代表菌群的相对丰度,X轴代表各种菌群;单星号表示差异显著,P<0.05; 双星号代表差异非常显著,P<0.01。
图3两组研究对象菌群组成结构的比较
3 讨 论
3.1结果分析 在菌群门的水平上,本研究中健康人群鼻前庭的主要优势菌门为变形菌门、厚壁菌门、放线菌门,与以往的研究结果相同,即健康人体鼻腔中主要的细菌门是放线菌门(50.7%~68.0%)、厚壁菌门(24.7%~27.0%)和变形菌门(4.0%~20.7%)[2,9]。尽管所占比例有所差异,这种差异可能受患者所处地理环境、人口水平和患者皮肤菌群的复杂程度影响,比如环境湿度就是影响放线菌门的一个重要外界因素[10]。
在菌群属的研究水平上,过敏性鼻炎患者与健康对照组之间有明显差异的菌属有13个,其中,过敏性鼻炎患者的黄单胞菌科中的一个未注释的菌属、棒状杆菌属、嗜胨菌属、厌氧球菌属、大芬戈尔德菌属、丙酸杆菌属、短杆菌属和考克菌属共8个菌属的丰度较健康受试者菌群明显减少。而过敏性鼻炎患者的希瓦菌属、微小单胞菌属、鞘脂单胞菌科中的一个未注释的菌属、嗜盐单胞菌和叶瘤菌属共5个菌属的丰度较健康受试者明显升高。棒状杆菌属一般出现在土壤、水源、植物和食品中,它们中的大部分是无害的,也没有致病性,但可利用非典型途径进入组织(伤口)或产生机会性致病[11]。非白喉棒状杆菌甚至可见于人和动物体内黏膜及皮肤表面[12]。嗜胨菌属是阴道和肠道菌群的一部分[13],其出现在糖尿病患者皮肤表面、软组织感染、关节感染、手术部位感染、绒毛膜羊膜炎和血液感染中[14]。厌氧球菌属可以导致人体不同部位的感染,如中耳炎、口腔感染、鼻窦炎和创伤后感染,此类菌在人体皮肤及黏膜腔内深处表面广泛存在。大芬戈尔德菌是一种机会性人体致病菌,通常定居在皮肤及黏膜表面[15],常见于慢性溃疡患者生物膜,如肥胖患者脚部及褥疮性溃疡[16]。研究表明,丙酸杆菌属是最流行的与人体皮肤相关的菌属[17]。主要为兼性寄生,或共生于人体及其他动物,多存在汗腺、皮脂腺或皮肤其他地方。实际上这些细菌无处不在,对大多数人来说不会造成问题,但与痤疮及某些皮肤问题有一定关联[18]。以上证据表明过敏性鼻炎较健康受试者较少的菌属大部分与人体皮肤和黏膜有关,都是人体的正常菌群,因此我们推测过敏性鼻炎与人体鼻腔的微生态失调相关。
3.2研究现状与创新性 研究发现,鼻腔微生物在过敏性鼻炎的发病过程中占有十分重要的地位。早在1989年,就有卫生假说表明,鼻腔病原体的暴露可能导致机体耐受性增高[19],儿童患过敏性鼻炎的速度明显降低。在2012年Heydenreich等报道某些G+菌能作为佐剂增强树突细胞成熟和炎症Th1、Th2和Th17的应答,提示微生物群落对过敏性鼻炎确实有影响[20]。还有学者提出慢性鼻窦炎中的大多数常见菌如肺炎链球菌、痤疮丙酸杆菌、棒状杆菌、流感嗜血杆菌和卡他莫拉菌可能与AR发病具有正相关性[5-6]。2014年Choi等用RFLP检测过敏性鼻炎患者鼻腔微生物群落的变化,结果表明,过敏性鼻炎患者的中鼻道内较健康对照组有更多种类的微生物,细菌多样性的升高与过敏性炎症反应相关[21]。但这些研究都依赖于传统的培养方式,而传统培养方式无法分析低丰度细菌及不能培养的细菌,因此无法反应鼻腔微生物的真实组成,且耗时长、工作量大。16S rRNA是原核生物核糖体16S亚基的基因的编码产物,存在于所有细菌基因中,占细菌总RNA的80%以上。其序列长短适中,既有保守区又有高变区,其中可变区的V3-V4区可作为菌群分类的标志[22]。高通量测序以其极低单碱基测序成本和超高数据产出量为特征,为基因组学和后基因组学研究领域带来了新的研究方法和解决方案。16S rDNA高通量测序技术可对数百万DNA分子同时测序,具有高准确性,高通量,高灵敏度和低运行成本等突出优势。因此本研究选择此方式进行鼻前庭菌群组成的研究,以期能更真实地反映过敏性鼻炎的鼻腔微生物的真实组成。
3.3研究意义 为了探索鼻前庭微生物的变化与过敏性鼻炎的关系,以期揭示鼻腔微生态与过敏性鼻炎的相关性,期望能为过敏性鼻炎的个性化诊疗提供帮助。
3.4不足之处 由于实验经费的限制,本课题样品的数量有限,但据此我们可以做出合理性推测,健康人体和过敏性鼻炎患者鼻前庭微生物群落存在差异,这些差异菌群值得我们进一步深度研究。
[1] Bousquet J, van Cauwenberge P, Khaltaer N. Allergic rhinitis and its impact on asthma[J]. J Allergy Clin Immunol, 2001, 108(5 Suppl): S147-334.
[2]Yan M, Pamp SJ, Fukuyama J, et al. Nasal microenvironments and interspecific interactions influence nasal microbiota complexity andS.aureuscarriage[J]. Cell Host Microbe, 2013, 14(6): 631-640.
[3]Norrman E, Nyström L, Jönsson E, et al. Prevalence and incidence of asthma and rhinoconjunctivitis in Swedish teenagers[J]. Allergy, 1998, 53(1): 28.
[4]Yang HJ, Lee SY, Suh DI, et al. The cohort for childhood origin of asthma and allergic diseases(COCOA) study: Design, rationale and methods[J]. BMC Pulmon Med, 2014, 14(1): 109.
[5]Feazel ML, Robertson CE, Ramakrishnan VR, et al. Microbiome complexity andStaphylococcusaureusin chronic rhinosinusitis[J]. Laryngoscope, 2012, 122(2): 467-472.
[6]Chalermwatanachai T, Velásquez LC, Bachert C. The microbiome of the upper airways: Focus on chronic rhinosinusitis[J]. World Allergy Organiz J, 2015, 8(1): 1-14.
[7]Edgar RC, Flyvbjerg H. Error filtering, pair assembly and error correction for next-generation sequencing reads[J]. Bioinformatics, 2015, 31(21): 3476.
[8]Greiner AN, Meltzer EO. Overview of the treatment of allergic rhinitis and nonallergic rhinopathy[J]. Proc Am Thoracic Society, 2011, 8(1): 121.
[9]Frank DN, Feazel LM, Bessesen MT, et al. The human nasal microbiota andStaphylococcusaureuscarriage[J]. PLoS One, 2010, 5(5): e10598.
[10] Rouadi P, Baroody FM, Abbott D, et al. A technique to measure the ability of the human nose to warm and humidify air[J]. J Appl Physiol, 1999, 87(1): 400-406.
[11] Sharma H, Tal R, Clark NA, et al. Microbiota and pelvic inflammatory disease[J]. Semin Reproduct Med, 2014, 32(1): 43.
[12] Brown K, Church D, Lynch T, et al. Bloodstream infections due toPeptoniphilusspp. : Report of 15 cases[J]. Clin Microbiol & Infect, 2014, 20(11): O857-O860.
[13] De MC, David CH, Provost B, et al. Finegoldia magna, not a well-known infectious agent of bacteriemic post-sternotomy mediastinitis[J]. Anaerobe, 2014, 32: 32-33.
[14] Lau SK, Woo PC, Fung AM, et al. Anaerobic, non-sporulating, Gram-positive bacilli bacteraemia characterized by 16S rRNA gene sequencing[J]. J Med Microbiol, 2004, 53(Pt 12): 1247.
[15] Cheng P, Song W, Gong X, et al. Proteomic approaches ofTrichodermahamatumto controlRalstoniasolanacearum causing pepper bacterial wilt[J]. Int J Agric & Biol, 2015, 17(6): 1101-1109.
[16] Bojar RA, Holland KT. Acne andPropionibacteriumacnes[J]. Clin Dermatol, 2004, 22(5): 375.
[17] Postollec F, Falentin H, Pavan S, et al. Recent advances in quantitative PCR(qPCR) applications in food microbiology[J]. Food Microbiol, 2011, 28(5): 848.
[19] Strachan DP. Hay fever, hygiene, and household size[J]. BMJ, 1989, 299(6710): 1259-1260.
[20] Heydenreich B, Bellinghausen I, König B, et al. Gram-positive bacteria on grass pollen exhibit adjuvant activity inducing inflammatory T cell responses[J]. Clin & Exp Allergy J Br Society Allergy & Clin Immunol, 2012, 42(1): 76.
[21] Caporaso JG, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data[J]. Nat Methods, 2010, 7(5): 335.
[22] HUANG Wenxian, WANG Heping, WANG Yulei, et al. High-throughput sequencing reveals the change of gut microbiota in infants with pneumonia following antibiotic treatment[J]. Chin J Microecol, 2016, 28(5): 497-500. (in Chinese)
黄文献, 王和平, 王玉蕾, 等. 高通量测序检测肺炎婴幼儿抗生素治疗前后肠道菌群变化[J]. 中国微生态学杂志, 2016, 28(5): 497-500.
猜你喜欢
基层中医药(2021年4期)2021-07-22 07:15:28
基层中医药(2020年9期)2020-11-27 01:55:10
家庭医学(下半月)(2020年4期)2020-05-30 12:42:42
家庭医学(下半月)(2020年4期)2020-05-30 12:42:42
家庭医学(下半月)(2020年4期)2020-05-30 12:42:40
中国比较医学杂志(2020年4期)2020-05-26 05:47:22
水生生物学报(2019年4期)2019-07-20 08:08:10
兽医导刊(2019年1期)2019-02-21 01:14:18
生物安全学报(2019年3期)2019-02-15 16:54:12
川北医学院学报(2019年6期)2019-02-10 10:48:32
