
食甲基杆菌J1-1吡咯喹啉醌生物合成突变株的筛选和鉴定
2018-04-09 03:37李彤景爱霞杨亚欣刘晓阳汪建华刘党生熊向华张惟材
生物技术通讯 2018年2期
李彤,景爱霞,杨亚欣,刘晓阳,汪建华,刘党生,熊向华,张惟材
1.沈阳药科大学,辽宁 沈阳 110016;2.军事科学院 军事医学研究院,北京 100071
吡咯喹啉醌(pyrroloquinoline quinine,PQQ)是20世纪70年代末发现的继烟酰胺核苷酸和黄素核苷酸后第三类氧化还原酶的辅酶[1],具有抗氧化、清除氧自由基、增强线粒体功能、保护心肌损伤、促进认知等多种生理功能[2-4]。PQQ的制备方法主要有化学合成法和生物合成法。化学合成步骤多、成本高、易造成环境污染,而生物合成周期短、成本低,被认为是最有前景的工业化生产路线。迄今发现只有某些革兰阴性菌能产生PQQ,如乙酸钙不动杆菌、肺炎克雷伯菌、扭脱甲基杆菌、氧化葡糖杆菌等[5]。目前PQQ合成基因以及PQQ的生物合成途径还不完全清楚。从革兰阴性菌分离到的PQQ合成基因包括pqqA、pqqB、pqqC、pqqD、pqqE、pqqF、pqqG,大多以基因簇的形式存在。其中pqqA基因编码的小肽PqqA含有PQQ生物合成的骨架物质谷氨酸和酪氨酸,被认为是PQQ合成的前体;PqqB可能与PQQ的跨膜运输有关;PqqC负责催化PQQ合成最后一步反应;PqqD的功能尚不清楚;PqqE的作用可能是连接前体中的谷氨酸和酪氨酸残基;PqqF、PqqG在PQQ合成中可能发挥内切酶的作用[6]。
本实验室从土壤中分离筛选到一株PQQ产生菌MP688,后经诱变得到高产菌株J1-1。已完成MP688的全基因组测序,发现其中PQQ合成基因包括基因簇pqqABCDE和pqqFG,以及4个单独拷贝的pqqA基因[7]。在此,我们通过Tn5转座诱变技术进一步筛选食甲基杆菌J1-1中参与PQQ生物合成的相关基因,为阐明PQQ的生物合成机制奠定基础。
1 材料与方法
1.1 材料
食甲基杆菌J1-1、大肠杆菌λ-pir感受态细胞、质粒pCM66和pGX由本实验室保存;大肠杆菌DH5α感受态细胞、快速质粒小提试剂盒、细菌基因组DNA提取试剂盒、琼脂糖凝胶DNA回收纯化试剂盒购自北京天根生化科技有限公司;ENTn5<R6Kγori/KAN-2>TNP Transposome Kit购自Epicentre公司;限制性内切酶购自Thermo公司;DNA聚合酶、pEASY-Uni SeamLess Cloning and Assembly Kit、T4DNA连接酶、DNA marker购自北京全式金生物技术有限公司;PCR引物(表1)合成及测序由北京六合华大基因科技股份有限公司完成。
LB培养基:胰蛋白胨10 g/L,酵母提取物5 g/L,NaCl 10 g/L,121℃灭菌20 min。
HC培养基:MgSO4·7H2O 0.2 g/L,KH2PO42.8 g/L,(NH4)2SO43.0 g/L,Na2HPO46.0 g/L,MnCl2· 4H2O 5.0 mg/L,ZnSO4·7H2O 5.0 mg/L,CuSO4· 5H2O 0.5 mg/L,柠檬酸铁60.0 mg/L,甲醇10 mL/ L,pH7.0,115℃灭菌30 min。
1.2 分子生物学操作
PCR、酶切、连接、转化等常规分子生物学操作参照文献方法进行[8]。
1.3 Tn5突变体库的构建
从 EN-Tn5<R6Kγori/KAN-2>TNP Transpo⁃some Kit中取1 μL Transposome加入J1-1感受态细胞中,冰浴5 min混匀后转移到预冷的电击杯中,2.5 kV电击后迅速加入500 μL HC培养基,30℃、200 r/min摇床振荡培养4 h,涂布于含50 μg/mL卡那霉素(Kan)的HC平板上,倒置培养2~3 d。挑取单菌落于5 mL HC Kan抗性试管中,30℃、200 r/min培养4 d,连续传3代后仍有Kan抗性,说明转座序列可以稳定传代,以此方法建立突变体库。

表1 PCR引物序列
1.4 PQQ合成缺陷突变株的筛选及PQQ合成水平检测
PQQ是一种棕红色小分子化合物,食甲基杆菌J1-1在发酵过程中,PQQ被逐渐分泌到胞外,使发酵液呈红色,发酵液颜色可直观反映PQQ合成水平。挑取Tn5转座突变体库上的单菌落于5 mL HC液体培养基中,以J1-1为对照,30℃、200 r/min振荡培养4 d,挑选发酵液颜色呈白色且与对照菌发酵液颜色差异较大的菌株作为初筛突变株,进一步采用分光光度法[9]检测发酵液中PQQ合成水平。
1.5 质粒拯救法鉴定插入位点
突变菌株基因组提取按照基因组提取试剂盒说明书进行。选择适当限制性内切酶单酶切突变株基因组,酶切产物经T4DNA连接酶连接,热激转化到大肠杆菌λ-pir感受态细胞中,涂布于含50 μg/mL Kan的LB抗性平板上,挑取单菌落37℃振荡培养,12 h后用质粒小提试剂盒提取质粒,由华大公司用引物SeFP/SeRP测序,测序结果通过NCBI的BLAST比对,确定Tn5插入位点。
1.6 mpq0056基因敲除
以J1-1基因组为模板,选用引物up1500-F/ up1500-R和down1500-F/down1500-R分别扩增上、下游同源臂基因;以质粒pGX-Gm为模板,选用引物Gm-F/Gm-R扩增庆大霉素(Gm)基因片段,扩增产物经琼脂糖凝胶电泳后,用胶回收试剂盒纯化。将上、下游同源臂基因,Gm基因和经XhoⅠ、NotⅠ双酶切的载体pGX-Gm用pEASYUni SeamLess Cloning and Assembly Kit重组转化,获得pGX-up-Gm-down重组质粒。
以pGX-up-Gm-down质粒为模板,选用引物up1500-F/down1500-R扩增片段-up-Gm-down-,纯化后电击转入J1-1感受态细胞,从HC抗性平板上挑取单菌落,通过组合PCR验证敲除菌。
1.7 mpq0056基因回补及过表达
用启动子预测软件BPROM预测mpq0056基因的启动子位置。以J1-1基因组为模板,用引物mpq0056-F/mpq0056-R扩增mpq0056启动子及编码基因,回收纯化DNA,用限制性内切酶XbaⅠ、KpnⅠ分别双酶切载体pCM66及目的DNA并胶回收纯化,酶切产物经T4DNA连接酶连接,将连接产物热激转化大肠杆菌DH5α感受态细胞,筛选阳性克隆,提取质粒酶切验证。
将质粒pCM66-0056分别电击转化野生菌J1-1和敲除菌J1-1Δ0056,构建过表达菌株和回补菌株。HC抗性平板培养2~3 d后,挑取单克隆于HC液体培养基中,提取质粒复转到大肠杆菌DH5α中,测序鉴定。
1.8 菌株生长评价
挑取野生菌J1-1、突变菌1-52、敲除菌J1-1Δ 0056、回补菌和过表达菌单菌落于5 mL HC液体培养基中,30℃活化24 h,分别按1%的接种量接种至100 mL HC液体培养基中,30℃同步培养,间隔12 h取样测定D600nm值,绘制生长曲线。
2 结果
2.1 Tn5转座突变体库构建
对J1-1进行Tn5转座诱变,通过Kan抗性筛选,构建得到库容量为1.0×105的突变体库。以突变株基因组为模板,用引物Kan-F/R、MOD-F/R PCR扩增Kan与转座序列,分别获得约800和1923 bp的特异性条带,与预期相符(图1),表明转座序列已插入受体菌株染色体基因组。
2.2 Tn5转座突变株的筛选和插入位点鉴定
经Tn5突变体库筛选获得1株PQQ产量显著降低的突变株1-52,该突变株发酵液接近白色(图2)。将该突变株基因组单酶切自连转化后,从Kan抗性平板上挑取单菌落于LB液体培养后提取质粒并测序,测序结果经BLAST后,确定插入基因为mpq0056(Chorismate lyase,ubiC)(图3)。
2.3 mpq0056基因敲除
2.3.1mpq0056基因敲除质粒的构建 采用引物up1500-F/up1500-R和down1500-F/down1500-R菌落PCR验证构建的敲除质粒pGX-up-Gm-down,均在1500 bp处出现特异性条带,与预期相符(图4)。将敲除质粒送北京六合华大公司测序,结果进一步证实重组质粒构建成功。
2.3.2mpq0056基因敲除菌的鉴定 选用引物up1700-F/Gm-R、Gm-F/down1700-R、Gm-F/Gm-R,以敲除菌基因组为模板进行组合PCR鉴定,应分别扩增出2500、2500、800 bp的片段,而以野生菌基因组为模板时应为阴性结果,结果与以上预期完全一致(图5),说明mpq0056基因敲除成功。
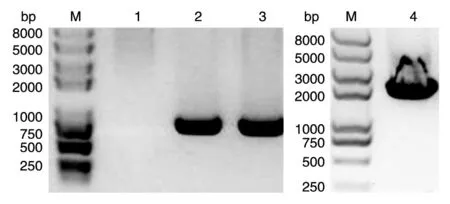
图1 缺陷株抗性基因及转座片段的PCR鉴定M:DNA marker;1:空白对照;2:阳性对照;3:1-52 PCR扩增Kan序列;4:1-52 PCR扩增转座序列

图2 野生菌J1-1和突变株1-52发酵液
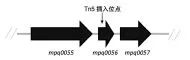
图3 1-52突变株Tn5插入位点

图4 pGX-up-Gm-down菌落PCR鉴定M:DNA marker;1:空白对照;2:阳性对照;3:mpq0056上游同源臂基因;4:mpq0056下游同源臂基因

图5 敲除菌J1-1Δ0056的组合PCR鉴定M:DNA marker;1:空白对照;2:野生菌(up1700-F/Gm-R);3:敲除菌(up1700-F/Gm-R);4:野生菌(Gm-F/down1700-R);5:敲除菌(Gm-F/down1700-R);6:野生菌(Gm-F/Gm-R);7:敲除菌(Gm-F/Gm-R)
2.4 mpq0056基因回补及过表达
用启动子预测软件BPROM预测结果显示在mpq0056基因上游200 bp内存在启动子序列(图6)。将mpq0056基因启动子区和编码区克隆到载体pCM66中,构建pCM66-0056载体,经XbaⅠ、KpnⅠ双酶切后出现2条DNA带,约8000 bp的条带与载体pCM66大小一致,约700 bp的条带与mpq0056的启动子和编码区片段大小一致(图7),表明pCM66-0056重组质粒构建成功。
提取回补菌株和过表达菌株质粒复转到大肠杆菌DH5α,提取质粒,用引物mpq0056-F/mpq0056-R测序,结果显示回补株和过表达菌株构建成功。
2.5 菌株表型鉴定
用分光光度法检测突变株1-52的生长及PQQ合成水平,突变株生长速度略慢,同时PQQ合成水平只有野生株的1/7~1/6。向突变株培养基中额外添加终浓度为10 mg/L的PQQ标准品(用“+”表示),突变株的生长基本恢复正常,但单位细胞的PQQ合成水平依然很低(图8)。表明突变基因mpq0056与PQQ的生物合成是有关联的。
敲除mpq0056基因后,敲除菌生长正常,单位细胞PQQ合成水平下降显著,与Tn5诱变得到的结果相符;回补及过表达菌株的生长均正常,回补基因mpq0056后,PQQ合成水平回复到野生菌水平(图9)。以上结果进一步说明基因mpq0056与PQQ的生物合成是相关的。
3 讨论
随着后基因组时代的到来,发现新基因和挖掘功能基因的方法越来越多,其中构建转座突变体库是最直接有效的方法之一。Tn5转座子是复合型转座子,在宿主中单拷贝复制。相比于常规的转座技术,Tn5具有突变性状稳定、突变基因容易定位、抗性筛选等优势[10],操作简单、转化效率高,能够快速获得大量稳定遗传的Tn5转座突变株。实践证明利用Tn5转座诱变筛选食甲基杆菌J1-1中PQQ合成基因的方法是可行的。前期研究发现,J1-1可利用的碳源并不多,其中以甲醇和果糖为碳源时菌体生长良好,当PQQ合成基因pqqBCDE敲除后,在果糖培养基中生长良好,却不能利用甲醇为碳源生长,当敲除菌中加入痕量的外源PQQ后菌体生长正常,这一现象说明菌体利用甲醇为碳源时是PQQ依赖的。本研究经Tn5转座诱变筛选PQQ合成基因是在以甲醇为碳源的平板上筛选,因以甲醇为碳源时PQQ合成水平高,表型明显,便于筛选,但目前这种方法只能筛选到PQQ合成水平下降的突变株,不合成PQQ突变株在甲醇为碳源的培养基中不生长。后续将尝试利用以果糖为碳源的平板进一步筛选PQQ合成基因。

图6 mpq0056上游前200 bp的启动子预测分析

图7 pCM66-0056双酶切鉴定M:DNA marker;1:pCM66-0056质粒XbaⅠ、KpnⅠ双酶切
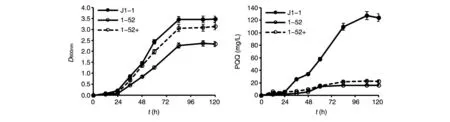
图8 突变株1-52的生长及PQQ合成水平检测
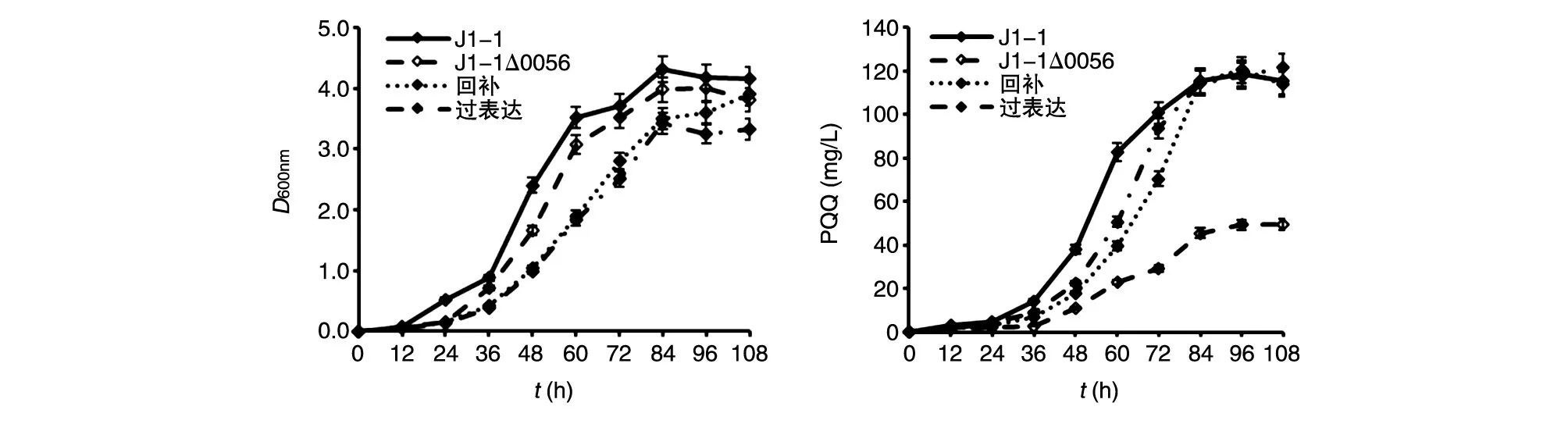
图9 mpq0056基因敲除、回补及过表达菌生长和PQQ合成水平检测
综上,经Tn5转座诱变获得一株插入位点为mpq0056的PQQ合成缺陷突变株,BLAST分析结果表明该基因编码产物可能是分支酸裂合酶,Nichols[11]等研究显示该酶在大肠杆菌中催化分支酸生成丙酮酸和对羟基苯甲酸,一方面作为泛醌类物质合成的前体,参与催化泛醌生物合成的第一步反应,另一方面作为芳香族氨基酸生物合成的中间代谢产物。通过基因组位置分析发现,mpq0056与上游基因mpq0055(ATP-dependent DNA helicase RecG)间隔区为73 bp,经在线启动子预测软件BPROM分析显示此间隔区存在启动子序列,提示mq0055与mpq0056基因不是同一基因簇,未影响上游基因功能;与下游基因mpq0057(4-hydroxybenzoate polyprenyl transferase,ubiA)间隔区为78 bp,功能注释存在相关性。我们推测食甲基杆菌J1-1中基因mpq0056影响PQQ生物合成的机制可能是通过影响芳香族氨基酸的代谢,间接影响PQQ前体物质合成。其影响机制须后续实验阐明。
[1] Duine J A,van der Meer R A,Groen B W.The co⁃factor pyrroloquinoline quinone[J].Annu Rev Nutr, 1990,10(10):297-318.
[2] 熊顺华,唐俊明,郭青平,等.吡咯喹啉醌D-半乳糖致大鼠皮层衰老的影响[J].中国现代医学杂志,2007,17 (15):1837-1840.
[3] Zhu B Q,Simonis U,Cecchini G,et al.Comparison of pyrroloquinoline quinone and/or metoprolol on myo⁃cardial infarct size and mitochondrial damage in a rat modelofischemia/reperfusion injury[J].JCardiovasc Pharmacol Ther,2006,11(2):119-128.
[4] Stites T,Storms D,Bauerly K,et al.Pyrroloquinoline quinone modulates mitochondrial quantity and function in mice[J].J Nutr,2006,136(2):390-396.
[5] Shen Y Q,Bonnot F,Imsand E M,et al.Distribution and properties of the genes encoding the biosynthesis ofthebacterialcofactor,pyrroloquinolinequinone[J]. Biochemistry,2012,51(11):2265-2275.
[6] Puehringer S,Metlitzky M,Schwarzenbacher R.The pyrroloquinoline quinone biosynthesis pathway revisit⁃ed:a structural approach[J].BMC Mol Biol,2008,9(8): 1-11.
[7] Xiong X H,Zhi J J,Yang L,et al.Complete genome sequence ofthe bacterium Methylovorus sp.strain MP688,a high-level producer of pyrroloquinolone qui⁃none[J].J Bacteriol,2011,193(4):1012-1013.
[8] 奥斯伯,布伦特,金斯顿,等.精编分子生物学实验指南[M].颜子颖,王海林,译.北京:科学出版社,1998.
[9] 杨延新,熊向华,游松,等.3种检测吡咯喹啉醌的方法比较[J].生物技术通讯,2011,21(4):554-557.
[10]年洪娟,陈丽梅,李昆志.Tn5转座突变技术在革兰氏阴性细菌分子遗传研究中的应用[J].中国生物工程杂志,2009,29(12):114-118.
[11]Nichols B P,Green J M.Cloning and sequencing of Escherichia coli ubiC and purification of chorismate ly⁃ase[J].J Bacteriol,1992,174(16):5309-5316.
猜你喜欢
环境工程技术学报(2022年3期)2022-06-05
中国土壤与肥料(2021年5期)2021-12-02
空间科学学报(2021年1期)2021-05-22
昆钢科技(2021年6期)2021-03-09
西南石油大学学报(自然科学版)(2019年5期)2019-12-20
天然产物研究与开发(2018年4期)2018-05-07
中成药(2018年1期)2018-02-02
环境保护与循环经济(2017年5期)2018-01-22
中国果菜(2016年9期)2016-03-01
电源技术(2016年9期)2016-02-27
